
Журнал «Здоровье ребенка» 2 (23) 2010
Вернуться к номеру
К вопросу о тяжелых комбинированных иммунодефицитах у детей
Авторы: Никонец Л.Д., Бельская Е.А., Островский И.М., Толстикова Е.А., Областная детская клиническая больница, г. Донецк, Донецкий национальный медицинский университет им. М. Горького
Рубрики: Педиатрия/Неонатология
Версия для печати
Тяжелые комбинированные иммунодефициты — это гетерогенная группа врожденных заболеваний, характеризующаяся глубоким блоком Т-клеточной дифференцировки и нарушением функции Т-клеток, возникающих вследствие мутаций в генах, отвечающих за пролиферацию лимфоцитов. Характерны тяжелые оппортунистические, бактериальные, вирусные, грибковые и БЦЖ-инфекции с первого года жизни, гипоплазия тимуса, периферических лимфоузлов, задержка физического развития, лимфоцитопения, гипогаммаглобулинемия. Радикальным методом лечения является трансплантация костного мозга. В статье представлено собственное наблюдение.
Тяжелые комбинированные иммунодефициты, дети.
Основные клинические проявления тяжелого комбинированного иммунодефицита ( severe combined immunodeficiency — SCID )
Комбинированный иммунодефицит — синдром, характеризующийся отсутствием Т-лимфоцитов и выраженным нарушением адаптивного иммунитета. SCID у человека впервые описан в 1950 году у швейцарских младенцев с лимфоцитопенией, умерших от инфекции в течение первого года жизни. В последующие годы установлено, что SCID включает множество синдромов, имеющих различную генетическую природу и различные типы наследования. Первой идентифицированной в 1972 году причиной развития SCID был дефицит аденозиндезаминазы, а природа дефекта, вызывающего развитие Х-сцепленной формы SCID , выявлена только в 1993 году. В последующие 11 лет определены молекулярные механизмы развития еще нескольких синдромов. В настоящее время известна генетическая природа более 15 форм SCID .
Для больных со SCID характерно раннее, в первые недели и месяцы жизни, начало клинических проявлений заболевания в виде гипоплазии лимфоидной ткани, упорной диареи, мальабсорбции, инфекций кожи и слизистых, прогрессирующего поражения респираторного тракта. Возбудителями инфекций являются бактерии, вирусы, грибы, условно-патогенные микроорганизмы. Цитомегаловирусная инфекция протекает в виде интерстициальной пневмонии, гепатита, энтеровирусы и аденовирус вызывают менингоэнцефалиты. Очень часто встречается кандидоз слизистых и кожи, онихомикоз. Характерно развитие регионарной и/или генерализованной БЦЖ. На фоне тяжелых инфекций развивается отставание в физическом и моторном развитии. Следует помнить, что даже при наличии SCID у младенцев не сразу развиваются все вышеперечисленные симптомы. В течение 2–6 месяцев они могут расти и развиваться нормально, особенно если прививка BCG не была сделана.
При лабораторном обследовании в большинстве случаев выявляются выраженная лимфоцитопения, гипогаммаглобулинемия и снижение пролиферативной активности лимфоцитов.
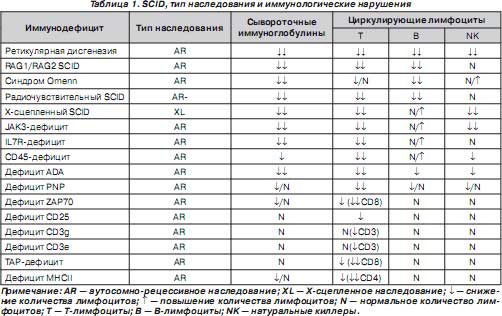
Основные известные в настоящее время формы SCID и их распределение представлены в табл. 1. Все эти синдромы отнесены к группе SCID [1–7], хотя при некоторых из них возможно позднее появление клинических проявлений иммунодефицита и довольно мягкое течение заболевания. Подобные случаи описаны при характеристике каждого из синдромов (табл. 1).
Молекулярно-генетические изменения и особенности иммунологических нарушений и клинического фенотипа некоторых отдельных форм SCID
Х-сцепленный SCID
Наиболее часто встречающаяся форма (более 50 % всех форм SCID ). Развивается в результате мутации гена общей g -цепи ( CD 132) интерлейкинов ( IL ) IL -2, IL -4, IL -7, IL -9, IL -15. Мутация g -цепи приводит к блокаде рецепторов, в результате чего клетки-мишени не способны ответить на действие соответствующих интерлейкинов.
IL -2 является одним из основных факторов роста лимфоцитов. При активации Т-клеток происходит экспрессия генов IL -2 и его рецептора ( IL -2 R ). Основными мишенями IL -2 являются активированные Т-, В- и NK -клетки. Главное действие, оказываемое им на Т-лимфоциты, — индукция пролиферации. IL -2 действует как один из ростовых факторов на В-лимфоциты. Это влияние усиливают IL -5 и интерферон гамма ( IFN - g ). IL -2 усиливает пролиферацию и цитотоксическую активность NK -клеток, расширяет спектр их цитотоксического действия.
Основными клетками-мишенями для IL -4 являются В-лимфоциты, для которых он является самым сильным ростовым фактором. IL -7 участвует в коммитировании развития клеток крови в сторону В-лимфоцитов, стимулирует развитие тимических предшественников Т-лимфоцитов, включает реаранжировку генов Т-клеточного рецептора ( TCR ), способствует выживанию Т-клеток, обусловливает антиген-независимое размножение Т-клеток вне тимуса. В отличие от большинства других цитокинов эффекты IL -7 не «дублируются» другими факторами. Удаление гена IL -7 приводит к опустошению тимуса, развитию тотальной лимфопении и тяжелого иммунодефицита.
Основным биологическим эффектом IL -15, продуцируемого макрофагами, является стимуляция пролиферации Т- и NK -клеток. Нарушение проведения сигнала с рецептора для IL -15 приводит к отсутствию NK -клеток. У больных наблюдается отсутствие Т-клеток, NK -клеток и повышение количества В-клеток. В результате отсутствия Т-клеточной регуляции продукция иммуноглобулинов В-клетками резко снижена. У пациента с Т – В + NK -лимфоцитарным фенотипом описаны с трехмесячного возраста диарея, респираторная инфекция Parainfluenza type 3, гипогаммаглобулинемия. У больного развились гепатомегалия, желтуха с лихорадкой, остеомиелит и лимфома. Мутационный анализ выявил инверсию ( IVS ) в общей гамма-цепи цитокиновых рецепторов у пациента и матери. В атипичных случаях у детей с X -сцепленным SCID , со средне-тяжелой картиной заболевания, возможно появление нормального количества циркулирующих зрелых Т-клеток и NK -клеток с частичной missens или splice - site мутациями. В Японии описан такой атипичный случай XSCID , представленный аутологичными Т- и NK -клетками и Omenn , — синдром с подобной манифестацией, включающей эритродермию, лимфаденопатию, гепатоспленомегалию, эозинофилию, низкий уровень IgG , повышенный уровень IgE , наличие активированных Т-клеток со средней рестрикцией Т-клеточного рецептора V b -репертуара.
В Турции частота встречаемости SCID — 1 : 10 000 живых новорожденных, 2,4 % от всех первичных иммунодефицитов. В 2001–2007 гг. зарегистрированы 27 пациентов со SCID . Преобладали девочки (16 девочек и 11 мальчиков). Высокая частота в кровно-родственных браках (22 пациента — 81 %). Средний возраст появления первых симптомов и установления диагноза SCID составлял 3,7 ± 2,7 и 7,3 ± 4,8 месяца соответственно. Рекуррентные синусо-пульмонарные инфекции (78 %), диарея (82 %), кандидоз кожи и слизистых (78 %) были частыми тяжелыми проявлениями SCID [4].
Мутации в генах, отвечающих за дифференцировку и пролиферацию лимфоцитов, могут приводить к мягким фенотипам, часто с проявлениями иммунной дисрегуляции, которые приводят к инфекциям. Описан пятилетний мальчик с гранулематозом кожи, нейтропенией, среднетяжелой лимфоцитопенией. Он имел 500/мл Т-клеток, 700/мл B -клеток и 10/мл NK -клеток с менее 10 % наивных CD 4+ клеток, преобладали гамма/дельта Т-клетки (50 %). Низкое количество Т- и NK -клеток с нормальным количеством B -клеток позволило диагностировать дефект в IL 2 R g общей цепи. При генетическом обследовании найдена точечная мутация в В-, NK - и буккальных эпителиальных клетках, хотя в a / b и g / d Т-клетках мутация отсутствовала [1–5].
Дефицит тирозинкиназы Janus ( Jak 3)
Тирозинкиназа семейства Janus — Jak 3 необходима для передачи активационного сигнала от общей гамма-цепи IL -2, 4, 7, 9, 15 к ядру клетки. Дефицит Jak 3 вызывает такие же глубокие нарушения дифференцировки Т- и NK -клеток, как и дефицит общей гамма-цепи. Иммунологические нарушения и клинические проявления у больных с дефицитом Jak 3 аналогичны таковым при X -сцепленной SCID [1, 6, 7].
Дефицит рецептора интерлейкина-7 ( IL -7 R )
Предшественники Т- и В-клеток экспрессируют функциональный IL -7 R , состоящий из альфа-цепи и общей гамма-цепи. Экспрессия этого рецептора критична для созревания Т-лимфоцитов, но не критична для развития В-лимфоцитов. Мутации гена альфа-цепи IL -7 R приводят к развитию SCID с фенотипом T – B + NK + и выраженным снижением концентраций сывороточных иммуноглобулинов [1, 2, 6, 7].
Принципы терапии SCID
SCID можно считать неотложным состоянием в педиатрии. Если SCID диагностирован в течение первого месяца жизни, адекватная терапия, в том числе проведение аллогенной HLA идентичной или гаплоидентичной трансплантации или пересадки гемпопоэтических стволовых клеток — hemopoetic stem cell tranplantation ( HSCT ) обеспечивает выживание 97 % пациентов независимо от формы иммунодефицита. В случае более поздней диагностики развиваются тяжелые инфекции, плохо поддающиеся терапии, и выживаемость пациентов резко падает.
Сразу после постановки диагноза SCID дети должны находиться в гнотобиологических условиях (стерильный бокс), в случае присоединения инфекций проводится интенсивная противомикробная, противовирусная и противогрибковая терапия, заместительная терапия внутривенным иммуноглобулином. Так как в Украине вакцинация BCG проводится в первые дни жизни, то дети со SCID в большинстве случаев оказываются инфицированными и у них развиваются BCG иты различной тяжести (от локальной до генерализованной инфекции). BCG -инфекция требует назначения длительной интенсивной противотуберкулезной терапии. Для профилактики пневмоцистных пневмоний назначают ко-тримоксазол. Особо отметим, что при необходимости проведения переливаний компонентов крови (эритроцитарная масса, тромбоконцентрат) следует использовать только облученные и отфильтрованные препараты. В случае переливания необлученных эритроцитов и тромбоцитов развивается фатальная посттрансфузионная реакция «трансплантат против хозяина» — GVHD ( Graft versus Host Disease ).
После проведения трансплантации клеток костного мозга или стволовых клеток не всегда развивается полная иммунологическая реконституция. У некоторых пациентов не происходит полного приживления В-клеток, и они нуждаются в пожизненной заместительной терапии внутривенным иммуноглобулином, однако это состояние не является несовместимым с жизнью иммунологическим дефектом [1–5, 7].
Приводим собственное наблюдение пациента с SCID . Мальчик от первой нормально протекавшей беременности, срочных родов с массой 3300,0 г , на естественном вскармливании. Мать здорова. У отца хронический фурункулез. Брак не родственный. Случаев смерти детей в родословной не наблюдалось. Привит БЦЖ. Голову держит с 3 месяцев, сидит с 6 месяцев. До 4 месяцев не болел. В 4 месяца — первый эпизод двусторонней пневмонии, дерматит, кандидоз ротовой полости, по поводу чего получал антибактериальные, противогрибковые препараты с выздоровлением. Поступил повторно в клинику в связи с рецидивом двусторонней пневмонии, с хронической диареей, рецидивирующим оральным кандидозом, гранулематозным поражением кожи.
В момент осмотра пациенту 7 месяцев. Масса 6100,0 г. Дефицит массы 20 %. Состояние тяжелое, обусловлено сердечно-легочной недостаточностью. Субфебрильно лихорадит. Подкожно-жировой слой отсутствует. Рубец после БЦЖ — 3 см . На голенях, бедрах, предплечьях, передней брюшной стенке — множественные подкожные гранулемы 3–5 мм в диаметре, плотные, цианотичной окраски. Большой родничок — 1,5 ´ 2,0 см . Периферические лимфоузлы не пальпируются. ЧД — 56 в мин. Одышка смешанного характера с участием вспомогательной мускулатуры в акте дыхания. Перкуторно над легкими — укорочение легочного звука в задне-нижних отделах. Дыхание жесткое, единичные мелкопузырчатые и крепитирующие хрипы в задне-нижних отделах. ЧСС — 136–148 в мин. Перкуторно границы сердца не расширены. Тоны сердца приглушены, тахикардия, систолический шум над предсердечной областью. Печень выступает из-под края реберной дуги на 1,5 см . Селезенка выступает из-под края реберной дуги на 1 см . Стул жидкий 4–5 раз в день, непереваренный, с комочками и слизью.
Данные дополнительного обследования
Общий анализ крови: Нв — 123 г/л; эр — 3,8 ´ 10 12 ; цп — 0,9; тромб. — 254,6 ´ 10 3 ; СОЭ — 6 мм/ч; Л — 8,1 ´ 10 9 ; э — 1; п — 4; с — 70; л — 15 (1350 кл/мл); м — 10.
СД3 + — 21 % — 379 кл/мл ( N — 58–67 % — 1900–3600); СД4 + — 9 % — 162 кл/мл ( N — 38–50 % — 1500–2800); СД8 + — 5 % — 90 кл/мл ( N — 18–25 % — 800–1200); СД20 + — 39,77 % — 718 кл/мл (N — 19–31 % — 500–1500); СД4/СД8 — 1,8 % ( N — 1,5/2,9).
IgG — 3,5 г/л (норма 6,61 ± 2,96 г/л), IgA — 0,2 г/л (норма 0,37 ± 0,18 г/л), IgM — 0,65 г/л (норма 0,54 ± ± 0,23 г/л)/ Кровь на ВИЧ — отрицательна.
Общий билирубин — 11,7 мкмоль/л за счет непрямого АЛТ — 0,17 мкк/л, ACT — 0,1 мкк/л, тимоловая проба — 3,7 Sh ед., кальций — 2,1 ммоль/л, фосфор — 1,61 ммоль/л, калий — 4,9 ммоль/л, натрий — 136,1 ммоль/л, амилаза — 9,2 мкк/л, мочевина — 4,7 ммоль/л, креатинин — 0,082 ммоль/л, общий белок — 43,9 г/л.
Бак. посев кала на кишечную группу: цитробактер 10 6 , чувствительный к норфлоксацину, цефотаксиму, цефтриаксону. Мазок из зева на грибы: Candida albicans 10 6 , чувствительная к нистатину, интраконазолу. Мазок из носа на грибы: отрицательный.
Кал на простейшие и яйца гельминтов: не обнаружены.
Копрограмма: переваримая клетчатка — много, нейтральный жир ++, лейкоциты — 15–20 п/зр.
Рентгенография органов грудной клетки: справа в проекции кардио-диафрагмального угла и над верхушкой правого легкого неоднородная инфильтрация легочной ткани. В прикорневой зоне единичные мелкоочаговые тени. Слева в 3-м межреберье в прикорневой зоне — неоднородная инфильтрация. Легочный рисунок усилен. Средостение не расширено. Сердце и синусы — норма. Заключение: двусторонняя пневмония.
ЭКГ: ЧСС 180 в минуту, полугоризонтальная электрическая позиция сердца, синусовая тахикардия, снижение вольтажа, изменения в миокарде.
Эхо-КГ: КДО — 21 мл, ФВ — 52 %, недостаточность митрального клапана 2-й ст., легочная гипертензия 1-й ст., Р в ЛА — 38 мм рт.ст., вегетаций на клапанах нет.
УЗИ органов брюшной полости. Печень: контуры ровные, четкие, структура повышенной эхогенности. Край печени выступает на 2 см по срединно-ключичной линии, эластичный. Экскурсия при дыхании 1 см . Диаметр воротной вены в месте пересечения с нижней полой 4 мм . Селезенка 57 ´ 24 мм , паренхима неоднородная за счет гипоэхогенных мишеневидных включений диаметром 5–7 мм. УЗИ тимуса: не лоцируется.
Бронхоскопия: нормальная эндоскопическая картина бронхолегочного дерева.
Диагноз: тяжелый комбинированный иммунодефицит Т – В + .
Терапия: внутривенный иммуноглобулин в суммарной дозе 800 мг, антибиотики — цефтриаксон в/в, амикацин в/в, ципрофлоксацин в/в, метрогил в/в, бактрим, рифампицин, флуконазол в/в, ацикловир, кардиотрофические препараты, сердечные гликозиды, мочегонные, муколитики, пробиотики, рибоксин и панангин в/в, зодак.
На фоне лечения уменьшилась степень дыхательной недостаточности, нормализовалась температура тела. В клинике находился 17 дней.
Ребенок направлен для уточнения фенотипа иммунодефицита ( X -сцепленный? JAK 3 дефицит? IL -7 R -дефицит?), генетического обследования и решения вопроса о трансплантации костного мозга в Западно-Украинский центр детской иммунологии, г. Львов.
1. Кондратенко И.В., Бологов А.А. Первичные иммунодефициты. — М, 2005. — С. 119-132.
2. Shchebet S.O., Mahis A.A., Aleshkevich S.N., Belevtsev M.O., Aleinikova O.V. Severe combined immunodeficiency disorder due to mutation in IL-7Rgene: a case report from Belarus // J. Clinical Experimental Immunology. — 2008. — V. 154, suppl. 1. — Р. 28.
3. Wada T.W., Yasui M., Toma T., Nakayama Y., Nishida M., Shimizu M., Okajima M., Kasahara Y., Koizumi S., Inoue M., Kawa K., Yachie A. Detection of revertant T lymphocytes in skin lesions of atypical x-linked severe combined immunodeficiency mimicking Omenn syndrome // J. Clinical Experimental Immunology. — 2008. — V. 154, suppl. 1. — Р. 32-33.
4. Reisli I., Artac H., Kara R., Van den Elsen P.J., Van der Burg M., van Dongen J.M. The spectrum of severe combined immunodeficiency in Konya, Turkey // J. Clinical Experimental Immunology. — 2008. — V. 154, suppl. 1. — Р. 39.
5. Speckmann C., Pannicke U., Nikolopoulos E., Shwarz K., Fish P., Friedrich W., Niehues T., Gilmour K., Buiting K., Schlesier M., Eibel H., Rohr J., Superti-Furga A., Grob-Wieltsch V., Ehl S. Somatic reversion in a patient with X-linked SCID a limited cure // J. Clinical Experimental Immunology. — 2008. — V. 154, suppl. 1. — Р. 42.
6. Bucley R.H. Primary immunodeficiency diseases due to defects of lyphocytes // New. Eng. J. Med. — 2000. — V. 343. — P. 1313-1324.
7. Bucley R.H. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution // Annu. Rev. Immunol. — 2004. — V. 55. — P. 625-656.