
Журнал «Здоровье ребенка» 2 (23) 2010
Вернуться к номеру
Молекулярно-генетические механизмы развития и современные методы лечения легочной артериальной гипертензии у детей 1. Генетическая предрасположенность
Авторы: Волосовец А.П., Национальный медицинский университет им. А. А. Богомольца, г. Киев; Абатуров А.Е., Днепропетровская государственная медицинская академия
Рубрики: Педиатрия/Неонатология
Версия для печати
В обзоре рассмотрены современные представления о генетической предрасположенности к легочной артериальной гипертензии. Развитие легочной артериальной гипертензии ассоциировано с мутациями генов рецепторов семейства TGF-b (рецептора II типа — BMPR2, рецептора I типа — ALK1), структурного сокомпонента рецепторов TGF-b эндоглина. В фенотипической реализации данных мутаций принимают участие множество других генов, в частности гены транспортера серотонина, вазоактивного интестинального пептида, активность которых определяет сократимость и скорость пролиферации гладкомышечных клеток сосудистых стенок.
Легочная артериальная гипертензия, молекулярно-генетические механизмы, дети.
Введение
Легочная артериальная гипертензия (ЛАГ) является редко встречающимся заболеванием, которое характеризуется фатальным исходом. Согласно данным французских и шотландских исследователей, распространенность первичной легочной гипертензии составляет от 15 до 26 случаев на 1 миллион населения [9, 30, 161].
Впервые ЛАГ, развитие которой не было связано с какими-либо причинами, протекающая с облитерацией мелких легочных сосудов и формированием правосторонней сердечной недостаточности, была описана E. Romberg в 1891 году [172].
ЛАГ — это патологическое состояние, характеризующееся прогрессирующим повышением сосудистого давления в легочной артерии в результате облитерирующего поражения мелких легочных сосудов, которое клинически проявляется снижением толерантности к физическим нагрузкам, инспираторной одышкой, обмороками, болью в области груди и приводит к развитию правожелудочковой сердечной недостаточности и преждевременной смерти [29, 132]. Поражение мелких легочных сосудов является патогномоничным признаком первичной легочной гипертензии, поражение крупных сосудов и дилатация правого желудочка сердца — вторичные компоненты заболевания. В процессе поражения мелких сосудов легочной ткани выделяют несколько стадий: 1) распространение гладкомышечных клеток в сосудистые стенки сосудов, которые в норме лишены мышечных клеток, преимущественно в области респираторных ацинусов за счет дифференцировки перицитов в зрелые гладкомышечные клетки; 2) мускуляризация (гипертрофия) мышечных проксимальных артерий, которая приводит к увеличению толщины медии и уменьшению диаметра просвета сосудов; 3) формирование из пролиферирующих и мигрирующих в субэндотелиальное пространство клеток и экстрацеллюлярного матрикса между эндотелием и внутренней эластической пластиной нового слоя клеток, получившего название неоинтима; 4) формирование плексиформных поражений [138, 140]. У новорожденных и детей раннего возраста ЛАГ обусловлена преимущественно недостаточностью легочной васкуляризации за счет уменьшения количества мелких артерий или увеличения их мускуляризации. У детей старшего возраста при ЛАГ наблюдаются как мускуляризация дистальных артерий, так и прогрессирующая гиперплазия интимы, плексиформные поражения легочных сосудов [163]. Повышение давления в легочной артерии более 25 мм рт.ст. в состоянии покоя и более 30 мм рт.ст. при физической нагрузке является диагностическим критерием ЛАГ [29, 31, 162].
Классификация
Венецианская классификация легочной артериальной гипертензии (2003) [31, 75].
1. Легочная артериальная гипертензия:
1.1. Идиопатическая (первичная) ЛАГ (ИЛАГ).
1.2. Семейная ЛАГ (СЛАГ).
1.3. Ассоциированная:
1.3.1. С системными заболеваниями соединительной ткани.
1.3.2. Врожденными пороками сердца (системно-легочные шунты).
1.3.3. Портальной гипертензией.
1.3.4. ВИЧ-инфекцией;
1.3.5. Лекарственными и токсическими воздействиями (ингибиторами аппетита (фенфлурамином, аминорексом, дексфенфлурамином), амфетаминами, L-триптофаном, кокаином, химиотерапевтическими препаратами, токсическими маслами, в частности, рапсовым).
1.3.6. другими (поражения щитовидной железы, болезнь Гошера, обменные болезни, наследственная геморрагическая телеангиэктазия, гемоглобинопатии, миелопролиферативные болезни, спленэктомия).
1.4. Ассоциированная со значительным поражением вен и капилляров.
1.5. Легочная веноокклюзионная болезнь.
1.6. Легочный капиллярный гемангиоматоз.
1.7. Персистирующая ЛАГ новорожденных.
2. ЛАГ, ассоциированная с поражениями левых отделов сердца:
2.1. Нарушение наполнения левого желудочка.
2.2. Поражения клапанного аппарата сердца (митральные пороки).
3. ЛАГ, ассоциированная с патологией дыхательной системы и/или гипоксемией:
3.1. Хроническая обструктивная болезнь легких.
3.2. Интерстициальные заболевания легких.
3.3. Нарушения дыхания во время сна.
3.4. Альвеолярная гиповентиляция.
3.5. Высокогорная ЛАГ.
3.6. Неонатальные поражения легких.
4. ЛАГ вследствие хронических тромботических или эмболических заболеваний:
4.1. Тромбоэмболическая обструкция проксимальных легочных артерий.
4.2. Тромбоэмболическая обструкция дистального русла легочной артерии.
4.3. Нетромботические легочные эмболии (опухоли, паразитарные заболевания, инородные тела).
5. Смешанные формы — саркоидоз, гистиоцитоз Х, лимфангиоматоз, компрессия легочных сосудов (аденопатия, опухоли, фиброзирующий медиастинит).
Генетическая предрасположенность
В настоящее время большинство исследователей считают, что СЛАГ и ИЛАГ являются генетически детерминированными заболеваниями, в основе которых лежат мутации генов двух рецепторов семейства трансформирующего фактора роста- b (TGF- b ) — рецептора II типа — костного морфогенетического белка (BMPR2), рецептора I типа — активин-подобной киназы (ALK1) (табл. 1) [19, 52, 88, 136.].

Суперсемейство TGF- b лигандов представляет большую группу (> 60) протеинов, являющихся полифункциональными факторами роста. Данное суперсемейство включает в себя три изоформы TGF- b (TGF- b 1 , TGF- b 2 и TGF- b 3 ), пять изоформ активинов, костные морфогенетические белки (BMP-1/толлоид, BMP-2, BMP-3/остеогенин, BMP-3 b /фактор роста и дифференцировки 10, BMP-5, BMP-6/ VG 1-связанная последовательность, BMP-7/остеогенный протеин-1, BMP-8/остеогенный протеин-1, BMP-8 b /остеогенный протеин-3, BMP-9/фактор роста и дифференцировки-2, BMP-10, BMP-11/фактор роста и дифференцировки-11, BMP-15/фактор роста и дифференцировки-9 b ) и микостатин [39, 46, 68, 86, 229]. Изоформы TGF - b и некоторые BMP ( BMP -2, BMP -4, BMP -6 и BMP -7) экспрессируются эндотелиоцитами и гладкомышечными клетками сосудов [28]. Димеры TGF- b в организме человека ассоциированы с ингибирующими протеинами — латент-ассоциированным протеином (LAP) и латентным TGF- b -связывающим протеином (LTBP). Изоформы TGF- b приобретают физиологическую активность только после разобщения с данными белками. Диссоциация комплексов TGF- b /LAP, TGF- b /LTBP происходит при кооперации TGF- b с рецептором инсулиноподобного фактора роста II , рецептором плазминогена урокиназного типа, плазмином, TSP, фибриноподобной пропротеиновой конвертазой. В отличие от TGF - b протеины BMP продуцируются в активной форме [20, 128, 145].
Протеины суперсемейства TGF - b взаимодействуют с двумя типами специфических трансмембранных рецепторных серин/треониновых киназ [15]. Различают 7 разновидностей рецепторов I типа (активин-подобные рецепторные киназы/ ALK ) и 5 разновидностей рецепторов II типа ( T b R - II : BMPR 2 , AMHR 2 , TGFR 2 , ActR 2 A , ActR 2 B ) [104, 128]. Рецептор BMPR 2 отличается от других T b R - II наличием длинной С-терминальной последовательности внутриклеточного домена [32]. Различные клетки легочной ткани характеризуются разной степенью плотности экспрессии BMPR 2 . Максимально высокая степень экспрессии BMPR 2 на поверхности клеточной мембраны наблюдается у эндотелиоцитов и менее интенсивная — на фибробластах и гладкомышечных клетках [16, 159].
Димерные лиганды суперсемейства TGF- b связываются с рецепторами обоих типов. Изотипы TGF- b связываются с рецепторами I типа (ALK-1 и ALK-5) и II типа, протеины BMP — с рецепторами I типа (ALK-1, ALK-2, ALK-3, ALK-6) и II типа (BMPR-II, ActR-II A , ActR-II B ), активины — с рецепторами IIA типа (ActRIIA) и IIB типа (ALK4). Взаимодействие лиганда с рецепторами ведет к организации рецепторного тетрамера. Формирование рецепторного тетрамера обусловливает фосфорилирование рецептора I типа рецептором II типа, что приводит к индукции активности киназы рецептора I типа. В дальнейшей внутриклеточной передаче сигнала участвуют протеины Smad. Идентифицировано восемь различных протеинов Smad, которые организуют три функциональных класса: рецептор-ассоциированные протеины Smad (R-Smad), комедиаторы Smad (cо-Smad) и ингибиторы Smad (I-Smad). Протеины R-Smad (Smad1, 2, 3, 5 и 8) после непосредственной активации киназами рецептора I типа формируют гетеромеры с cо-Smad (Smad4). Гетеромерный комплекс R-Smad/Smad4 транслоцируется в ядро клетки и связывается с энхансерами специфических генов, изменяя их транскрипционную активность [86, 127, 186, 230]. Рецептор BMPR 2 взаимодействует с BMP -2, BMP -4, BMP -6 и BMP -7 [32]. BMP возбуждают определенные R - Smads — Smad 1, 5 и 8. При возбуждении Smad -пути, вызванном изотипами TGF - b , формируются комплексы Smad 2,3/ Smad 4, Smad 1/ Smad 4; а при индукции BMP — Smad 5/ Smad 4, Smad 8/ Smad 4 [127, 186]. Естественными ингибиторами внутриклеточного сигнального пути, который активируется BMP, являются ноггин, хордин, фоллистатин, BAMBI и Smurf-1 [38, 132]. В эндотелиоцитах, гладкомышечных клетках, фибробластах протеины Smad1/5, по всей вероятности, конкурируя за связь с Smad4, противодействуют передаче сигналов Smad2/3, что предопределяет антагонизм между TGF- b и BMP сигнальными путями [18, 22].
Низкие концентрации TGF - b через ALK -1 стимулируют ангиогенез, пролиферацию и миграцию эндотелиоцитов, а высокие концентрации TGF - b через ALK -5 ингибируют данные процессы и индуцируют экспрессию фибронектина, ингибитора активатора плазминогена-1 [86]. Влияние TGF - b на гладкомышечные клетки сосудов связано преимущественно с активацией ALK-5/T b R-II комплекса, который индуцирует их пролиферацию и экспрессию ими a -актина, миозина. Изоформы TGF- b обусловливают усиление синтеза клаудина-5, снижая парацеллюлярную проницаемость, и ингибируют синтез молекул адгезии, IL-6, макрофагальный хемоаттрактантный протеин MCP1/CCL2 [7, 71, 205, 231]. Такие BMP, как BMP-2, BMP-4, BMP-6 и BMP-7, оказывают действие на эндотелиоциты и гладкомышечные клетки, ингибируя не только их пролиферацию, но и предупреждая развитие апоптоза [140]. BMP -2 стимулирует миграцию эндотелиоцитов и ангиогенез, а BMP -4 индуцирует продукцию активных кислородосодержащих метаболитов [20].
В 2000 году впервые были представлены доказательства, свидетельствующие о ключевой роли BMPR2 в развитии легочной гипертензии. Была установлена ассоциация легочной гипертензии с мутациями гена BMPR2 [71, 92]. И в настоящее время большинством исследователей мутация данного гена рассматривается как основной генетический фактор, предопределяющий развитие ЛАГ [38, 131, 171, 201]. Ген BMPR2, расположенный на длинном плече хромосомы 2 (2q33), имеет 13 экзонов. Экзоны 1–3 кодируют экстрацеллюлярный домен, экзон 4 кодирует трансмембранный домен, экзоны 5–11 — серин/треониновый киназный домен, экзоны 12 и 13 — длинный интрацеллюлярный С-терминальный домен. Мутации гена BMPR2 у больных с СЛАГ обнаружены практически во всех экзонах, за исключением 5 и 13 [83, 201].
Мутации гена BMPR 2 встречаются в 30 % случаев всех форм ЛАГ [138], выявляются приблизительно в 70 % случаях СЛАГ [136] и в 11–40 % спорадических случаев легочной гипертензии [132, 201]. В настоящее время идентифицированы 144 различные мутации гена BMPR2 у больных СЛАГ [136, 176]. СЛАГ характеризуется аутосомно-доминантным типом наследования с вариабельной пенетрантностью [83]. Мутации гена
BMPR 2 сопровождаются снижением активности фосфориляции Smad 5, Smad 8, что ведет к растормаживанию TGF - b сигнального пути, усилению активации фактора транскрипции STAT 3, предопределяя гиперпролиферацию и развитие апоптозрезистентности у эндотелиоцитов (рис. 1) [18, 22]. Возбуждение аномального BMPR 2 сопровождается экспрессией тенасцина- C , который способствует пролиферации гладкомышечных клеток (рис. 1) [33, 204].
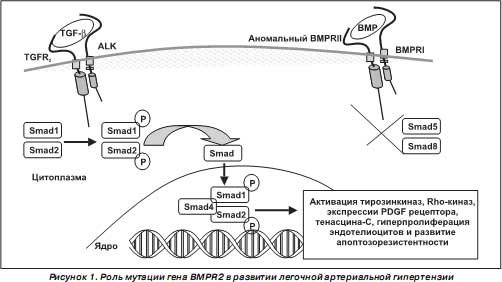
Однако последние исследования показали, что мутации гена BMPR 2, возможно, не являются критическим молекулярно-генетическим основанием для развития ЛАГ [82]. Только у 20 % людей с мутацией гена BMPR2 развивается клинически значимая ЛАГ [138, 232].
Мутации гена рецептора I типа ALK -1, который располагается на хромосоме 12 (12q11-q14), и гена эндоглина/CD105/ P 4 A 4 ( TGF - b -связывающего гликопротеина), который располагается на хромосоме 9 (9 q 33- q 34.1), обнаружены у лиц с сочетанной патологией — с наследственной геморрагической телеангиэктазией (прежнее название — синдром Рендю — Ослера — Вебера) и легочной гипертензией. Гомодимер эндоглин (180 кД) является трансмембранным гликопротеином I типа эндотелиоцитов, активированных макрофагов, фибробластов и гладкомышечных клеток, который входит в состав рецепторов семейства TGF- b и участвует в связывании TGF- b 1 , TGF- b 3 , активина А, BMP-2, BMP-7 [54, 228]. Мутации данных генов ведут к изменениям в активации Smad , которые характерны и для мутации гена BMPR 2 [52, 83, 140].
Считают, что для реализации мутаций генов BMPR 2, ALK -1 и эндоглина необходимо наличие других генетических или экологических факторов риска развития ЛАГ (табл. 2) [135].
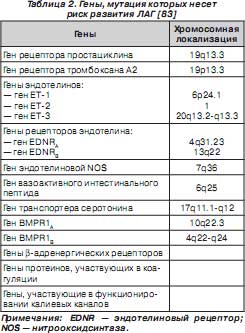
Таким образом, ЛАГ — генетически детерминированное заболевание, ассоциированное с мутациями генов рецепторов семейства TGF- b (рецептора II типа — BMPR2, рецептора I типа — ALK1), структурного сокомпонента рецепторов TGF- b эндоглина. В фенотипической реализации данных мутаций принимают участие множество других генов, в частности генов транспортера серотонина, вазоактивного интестинального пептида, активность которых определяет сократимость и скорость пролиферации гладкомышечных клеток сосудистых стенок.
Список литературы находится в редакции