
Международный неврологический журнал 4(4) 2005
Вернуться к номеру
Аномалии головного мозга (миграционные нарушения) у детей: клинико-радиологические проявления
Авторы: Е.П. Шестова, С.К. Евтушенко, Е.М. Соловьева, А.В. Душацкая
Рубрики: Неврология
Разделы: Справочник специалиста
Версия для печати
дети, аномалии головного мозга, миграционные нарушения, кортикальные дисплазии, нарушения архитектоники мозга.
Активное внедрение в практику детского невролога современных нейровизуализирующих методов обследования (КТ, МРТ головного мозга) позволило значительно расширить знания в области аномалий нейроонтогенетического процесса, определить их роль при оценке неврологического статуса ребенка и прогноза заболевания. Использование КТ и МРТ перевело прижизненное изучение структуры мозга на более высокую ступень, переключив познание с формального уровня «гипоксическая энцефалопатия» на уровень болезней, вызванных генетическим нарушением транскрипционных факторов [2].
Стало очевидным, что аномалии развития головного мозга являются наиболее частой причиной детской неврологической инвалидности. «Поломки» нейроонтогенетического процесса в большинстве своем представляют мультифакториальную патологию эмбрионального периода. Их моногенное наследование наблюдается нечасто, не более чем в 1% случаев. Большая часть врожденных пороков нервной системы формируется под воздействием повреждающих агентов в критические периоды эмбрионального развития органов и систем (по П.Г. Светлову), причем характер, вид порока зависит не от природы повреждающего агента (мутантный ген, химические мутагены, ионизирующая радиация, вирусы), а от возраста эмбриона. В связи с этим окончательно выяснить причину остановки развития органа или системы часто не представляется возможным.
Согласно современным представлениям о нарушении эмбриогенеза, на процессы формирования нервной системы могут повлиять следующие факторы:
— экзогенные токсины;
— генетические причины (преобладают спорадические случаи над наследуемыми);
— эндогенные токсины (метаболические нарушения у матери, в т.ч. повышение температуры материнского организма);
— инфекционные возбудители (цитомегаловирус, токсоплазма, листерии и др.).
Среди перечисленных причин, способствующих возникновению пороков развития, наиболее влиятельным является экзогенный фактор. В настоящее время бесспорным является факт глобального загрязнения среды обитания человека токсическими химическими веществами, что привело к значительному накоплению их в биосфере и дальнейшему поступлению в организм с продуктами питания, водой и воздухом. Количественный объем вновь возникающих мутаций может увеличиваться под влиянием мутагенных факторов среды, особенно таких, как ионизирующая радиация, активные химические соединения, некоторые биологические факторы. Это создает реальные предпосылки и условия для возникновения экологических поражений населения, проявляющихся прежде всего в эмбриотоксическом эффекте. Ускорение научно-технического прогресса принципиально изменило среду обитания человека. Во-первых, в ней появилось много факторов, с которыми человек ранее не сталкивался (например, 60 тыс. новых химических веществ). Во-вторых, среда изменяется в очень быстром темпе. Генотип популяций не успевает адекватно реагировать на изменение среды. Это приводит к тому, что в измененных экологических условиях возникают наследственные болезни нового класса — экогенетические болезни. Суть их сводится к тому, что определенная доля популяции имеет аллель, который проявляет патологическое действие при воздействии конкретного фактора среды. Новый класс экогенетических болезней до сих пор во многом еще не расшифрован, хотя он имеет огромное значение с точки зрения медицины труда, гигиены питания, окружающей среды [2]. Таким образом, увеличение числа пороков головного мозга вследствие спонтанных мутаций за счет пагубного влияния экологии все больше завоевывает место во взглядах на этиологию аномалий головного мозга.
Ведущая роль в диагностике многих пороков развития головного мозга принадлежит МРТ. С помощью этого обследования появилась возможность прижизненно выявить у больного ребенка такие аномалии мозга, которые ранее не определялись и соответственно не рассматривались в качестве причины неврологических расстройств у детей.
Особую ценность этот метод исследования приобретает в ранние сроки постнатального развития, так как в период новорожденности нарушенное состояние ребенка с пороком головного мозга необоснованно трактуется как внутриутробный менингоэнцефалит, гипоксическое или родовое повреждение нервной системы.
С помощью радиологических методов исследования четче визуализируются аномалии развития головного мозга, связанные с нарушением формы и строения (архитектоники) серого вещества. В меньшей степени это касается нарушения строения белого вещества. Поэтому большую часть пороков связывают с аномалией коры головного мозга и называют обобщенно корковыми дисплазиями.
Пороки головного мозга могут развиться на всех этапах эмбрионального и частично в фетальном периоде. Если «поломка» происходит во время формирования прозэнцефалона (2-3-й месяц внутриутробного развития), то возникают грубые, нередко «видимые глазом» пороки, о которых можно судить по наличию у больного сопутствующего лицевого дизостоза. К ним, например, относится голопрозэнцефалия, формирующаяся в строго фиксированные для ее развития сроки — 22-24-й дни внутриутробного развития. Такие аномалии головного мозга можно диагностировать и без дополнительных методов обследования, так как они сопряжены с грубой специфической неврологической симптоматикой, лицевым дизостозом, костными нарушениями.
Наибольший интерес для клинициста представляют пороки развития головного мозга, формирующиеся вследствие аномальных процессов развития мозга с 6-й по 20-ю неделю внутриутробного развития, происходящих в следующие периоды [8]:
— 3-4-й месяц гестации — процесс пролиферации (начало с 7-й недели);
— 3-5-й месяц гестации — центробежная миграция (начало с 8-й недели);
— 5-й месяц гестации — нейронная организация (ламинация, гиритация и сулькация).
Аномалии развития, формирующиеся в эти периоды, получили названия дисмиграционных нарушений головного мозга, или аномалий (болезней) нейронной миграции, трансмантийных дисплазий, нарушений архитектоники головного мозга и т.д. Часто эти названия и понятия смешиваются друг с другом, и порой их трудно разделить между собой, так как для каждого нарушения отсутствует специфическая симптоматика.
Исследователями обнаружена «мутация гена, нарушающая миграцию и расслоение нейронов при закладке структур мозга (RELN-ген, хромосома 7q22). Идентифицирован продукт этого гена — белок реелин-гликопротеид, служащий «проводником» для нейронов» [3].
Дисмиграционные нарушения проявляются симптомами средней и тяжелой степени органического поражения нервной системы. Их симптоматика во многом зависит от локализации нарушений структуры мозга и от их распространенности. Тем не менее, картинки на МРТ с явной аномалией головного мозга не всегда связаны с клиническими проявлениями патологических симптомов, что в настоящее время малообъяснимо. Вероятно, такие случаи являются исключением из правил, которые своим существованием и подтверждают эти правила. На степень неврологических расстройств может повлиять распространенность (площадь, объем) нарушений архитектоники головного мозга, известных в морфологии как мальпозиция и мальориентация. К сожалению, эти изменения плохо визуализируются на МРТ.
В целом симптоматика аномалий головного мозга четко выражена, но малоспецифична. Как правило, более грубые расстройства проявляются уже в период новорожденности нарушением адаптации, судорожным синдромом. По мере роста ребенка все более четко формируется задержка психомоторного развития, очаговый неврологический дефицит разной степени выраженности, часто — синдромокомплекс церебрального паралича. Эпилептические приступы у ребенка появляются в разные возрастные периоды, но чаще это происходит в первое десятилетие, нередко они резистентны к терапии [5]. Часто многие синдромы сочетаются друг с другом, но могут быть и изолированным проявлением порока развития головного мозга.
На возраст появления эпилептических приступов вследствие аномалий развития головного мозга может повлиять так называемый критический период развития ребенка. Не все тут ясно и понятно, но то, что эпиприступы появляются в различные возрастные периоды с наглядной визуализацией очага на МРТ, очевидно.
Почему эпиприступы, обусловленные пороками развития головного мозга, появляются в различные возрастные периоды? Ответ на этот вопрос частично может дать следующее утверждение Ю.Е. Вельтищева: «Изменения в генетической программе регуляции нормального роста и развития ребенка проявляются дискретностью, т.е. условной обособленностью отдельных периодов онтогенеза. …В постнатальном развитии критические периоды, или фазы, разделяют отдельные переломные этапы онтогенеза — они представляют собой своеобразные «верстовые столбы» в общей программе развития человека» [4]. Выделение постнатальных критических периодов, или фаз, развития было предложено Ю.Е. Вельтищевым и соавт. еще в 1983 г. При этом авторы основывались на объективных и хорошо известных проявлениях репрессии генов и генного переключения. К таким критическим периодам ребенка относятся:
— неонатальный;
— период 3-6 мес.;
— второй год жизни;
— возраст 6 лет;
— пубертатный период.
Понятие кортикальной дисплазии объединяет следующие варианты нарушения нейронной организации: лиссэнцефалия (агирия), пахигирия, микрополигирия, шизэнцефалия и трансмантийная дисплазия. Все они могут носить очаговый и генерализованный характер.
Одним из проявлений аномальной нейрональной и глиальной организации является трансмантийная дисплазия. По определению A.J. Barcovich, фокальная трансмантийная дисплазия (focal transmantle dysplasia) — это участок нарушения архитектоники, который образовался вследствие аномального развития стволовой клетки и расположен от стенки желудочка мозга до кортекса [5]. В литературе встречается такое понятие, как генерализованная фокальная трансмантийная дисплазия — большая область мозга с широкими нерегулярными извилинами и бороздами, с областями гиперинтенсивности и изоинтенсивности серого вещества к белому. Белое вещество, возможно, также аномально. Клинически трансмантийная дисплазия проявляется грубым очаговым неврологическим дефектом и эпилепсией.
Лиссэнцефалия (агирия) и пахигирия — недоразвитие мозговых извилин с гладкой поверхностью мозговых гемисфер, может быть тотальной и очаговой. В целом для лиссэнцефалии характерна умственная отсталость, раннее начало эпилепсии по типу инфантильных спазмов. Встречается как у девочек, так и у мальчиков. Часто регистрируется специфический паттерн ЭЭГ— генерализованная бета-активность высокой амплитуды. Тотальная агирия сопровождается ленточной гетеротопией, известной как синдром «двойной коры».
Описаны два морфологических типа лиссэнцефалии [7].
1-й — тип Bilschowski, для которого характерна 4-слойная кора. Четвертый слой сформирован из гетеротопических нейронов. Этот тип часто ассоциируется с другими аномалиями — гетеротопиями, макро- и микрогириями, шизэнцефалией и др. Клинически у больных отмечается гипотония, умственная отсталость, эпилептические пароксизмы по типу инфантильных спазмов, миоклоний, синдрома Леннокса — Гасто. Данный тип имеет генетическую и хромосомную детерминированность. Он является основным морфологическим признаком синдромов Варбурга и Секкеля (карликовость с птицеголовостью), синдромов Miller — Dilker и Norman — Roberts (эпилепсия, умственная отсталость, лицевой дисморфизм и другие стигмы), связанные с делецией 17-й хромосомы.
2-й тип — Walker's лиссэнцефалия с полным отсутствием кортикального слоя. Сочетается с гипоплазией мозжечка, моста, аномалией глаз и другими мальформациями мозга. Может встречаться при синдроме Dendy, синдроме Walker — Warburg.
Микрогирия (микрополигирия) — множество мелких, коротких, неглубоких извилин [7]. Чаще встречается фокальная микрогирия различной площади. Она может являться структурной основой многих генетических и хромосомных синдромов (Денди — Уокера, Арнольда —Чиари, Цельвегера, неонатальной адренолейкодистрофии и др.). Микрогирия является морфологическим дефектом при синдроме Foix — Chavany — Marie (умственная отсталость и псевдобульбарный паралич).
Микрогирия (полимикрогирия) — еще один вариант корковой дисплазии, обозначающий участок множества мелких, неглубоких извилин с нарушением строения серого вещества. Полимикрогирия, которая располагается с обеих сторон сильвиевой борозды, имеет специфические клинические проявления и получила название «врожденный двусторонний перисильвиев синдром» [7]. Клиническими симптомами ее являются: врожденная центральная диплегия лицевой, глоточной и жевательной мускулатуры, 100%-ное нарушение движения языка в сочетании с умственной отсталостью и эпилепсией. Судороги дебютируют, как правило, на первом году жизни. По своему характеру они могут быть как фокальными, так и генерализованными, иногда по типу инфантильных спазмов, резистентны к противосудорожной терапии.
В литературе можно встретить еще один термин — «фокальная корковая дисплазия» (ФКД), претендующий на самостоятельность. Но, по мнению многих исследователей [1, 5], это есть не что иное, как фокальная микрополигирия. ФКД — частичное нарушение нейроонтогенетических процессов нейронной миграции, результатом чего является образование патологических корковых участков (гигантские нейроны и причудливой формы астроциты, явления мальпозиции и мальориентации). Область преимущественной локализации ФКД — лобные и височные отделы мозга.
Основные нозологические критерии пароксизмов:
— эпиприпадки кратковременны (не более 1 мин);
— сложные парциальные припадки с минимальными явлениями постприступной спутанности;
— вторичная генерализация припадков происходит быстрее, чем при височной эпилепсии;
— часто демонстративные и необычные двигательные феномены;
— высокая частота автоматизмов в начальной фазе припадков;
— частые внезапные падения.
Для фокальной корковой дисплазии характерны выраженные, демонстративные и порой необычные двигательные феномены (жестовые автоматизмы (de novo), педалирование по типу топтания на месте), сопровождающие припадки. Во время приступа выражена моторная манифестация, включающая атипичные позные установки по типу билатеральных либо унилатеральных тонических поз и/или атонические эпизоды. Между приступами на ЭЭГ иногда отмечаются необычные и черезвычайно активные фокальные эпилептические разряды в виде повторяющихся спайков.
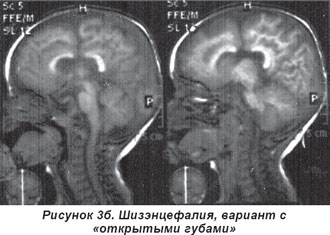
Своеобразной аномалией нейронной миграции является шизэнцефалия — тотальная патология с формированием глиальных миграционных траекторий, простирающаяся от желудочков до коры головного мозга. Данный порок развития хорошо визуализируется на томограммах головного мозга в виде различной степени выраженности щелей. В классическом варианте эти щели в коре головного мозга заканчиваются «открытыми губами» (рис. 3б). Стенки щелей выстланы патологически утолщенной корой. При данном пороке ликвородинамика не нарушена, она полностью компенсирована. Возле щелей, как правило, находятся очаги гетеротопии и/или полимикрогирии [7]. Клинические проявления шизэнцефалии ассоциируются со многими неврологическими симптомами и синдромами в виде:
— гемиплегии (при унилатеральном расположении);
— тетрапареза (при билатеральном расположении);
— судорожного синдрома;
— грубой задержки психомоторного развития.
Наиболее частым вариантом миграционных нарушений является гетеротопия — скопление нейронов, остановившихся в различных аномальных местах на пути следования к коре головного мозга. Такая остановка происходит не позже 5-го месяца внутриутробного развития. Изолированный участок узловатой массы называется «гетеротопион». В настоящее время описаны следующие варианты гетеротопии:
— субэпендимальная нодулярная (узелковая) гетеротопия;
— ленточная (слоистая, ламинарная) гетеротопия;
— изолированная (одиночная) гетеротопия;
— синдром «двойной коры».
Субэпендимальная узелковая (нодулярная) гетеротопия связана с мутацией гена FLN1 (Хq28). При этом мальчики погибают, а девочки рождаются с нодулярной гетеротопией. Субэпендимальная гетеротопия может быть одиночной и множественной. Локализуется чаще в области вентрикулярного треугольника и височных и затылочных рогах желудочков головного мозга. Нередко визуализируемые очаги субэпендимальной гетеротопии расценивают как очаговые ишемические поражения мозга или как кальцинаты, что затрудняет диагностику заболевания. У больных с изолированной субэпендимальной гетеротопией судороги обычно появляются во втором десятилетии. При локализации гетеротопиона в субкортикальной области кора головного мозга часто аномальная, с тонкими и мелкими извилинами. У таких больных отмечается различная степень задержки психомоторного развития, зависящаяся от размера и местоположения гетеротопиона. Пароксизмы развиваются почти у всех больных.
Наиболее часто у больных встречаются одиночные гетеротопионы. В отличие от гамартомы, при туберозном склерозе они не накапливают контрастное вещество.
Для ленточной (слоистой, ламинарной) гетеротопии характерно скопление гетеротопионов параллельно предполагаемой коре головного мозга. Данный вариант гетеротопии получил название синдрома «двойной коры». Его можно обнаружить при X-сцепленной лиссэнцефалии, развивающейся в результате мутации гена DCX (XLIS) Xq22.3-q23). При этом девочки рождаются с синдромом «двойной коры».
Представляем клинико-радиологический анализ 22 детей в возрасте от 2 месяцев до 1 года, находившихся в неврологическом отделении областной детской клинической больницы г. Донецка с 2003 по 2005 год.
Для анализа были отобраны больные со среднетяжелыми и тяжелыми расстройствами нервной системы, которым было проведено МРТ головного мозга в режимах Т1 и Т2.
У всех больных отмечали задержку психомоторного развития средней и тяжелой степени, отсутствие редукции тонических рефлексов, задержку формирования рефлексов выпрямления различной степени выраженности, полиморфные эпилептические приступы. Судороги были представлены синдромами Ohtahara и West, генерализованными тоническими пароксизмами, очаговыми и генерализованными миоклониями. В неонатальном периоде у 86% больных был диагностирован синдром угнетения, у 32% — синдром возбуждения, у 14% — судорожный синдром. У 65% детей эти состояния новорожденных были расценены только как гипоксическое поражение головного мозга, а у 12% — только как проявление внутриутробной инфекции. Ни у одного ребенка в период новорожденности не было проведено радиологическое исследование и не был диагностирован порок головного мозга.
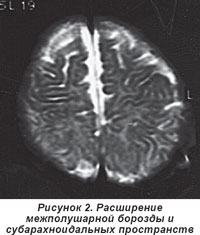
Наиболее частым признаком грубых расстройств нервной системы были фокальные трансмантийные пороки развития. На МРТ и КТ у этих детей находили прямые и косвенные признаки нарушения миграции нейронов, различное их сочетание. К косвенным признакам пороков головного мозга относили увеличение в размерах межполушарной борозды, субарахноидальных пространств, вентрикуломегалию без клинических проявлений гидроцефалии (рис. 1, 2). Косвенные признаки сопровождали практически все грубые дисмиграционные нарушения. Выраженное расширение межполушарной борозды и субарахноидальных пространств (как показатель недоразвития мозга) чаще обнаруживали в передних отделах полушарий.
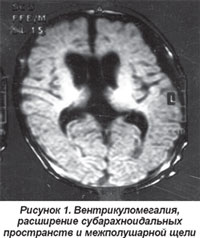
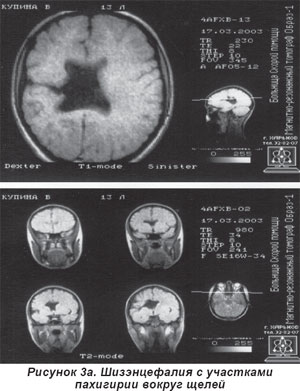
Среди прямых признаков аномалий мозга у больных с тяжелыми расстройствами нервной системы наиболее часто встречались расщелины мозга, расцененные нами как вариант шизэнцефалии (рис. 3а, 3б). Они были различных размеров, с одной или с обеих сторон.
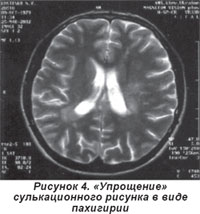
Еще одним частым прямым признаком аномалий головного мозга были различные нарушения рисунка извилин (сулькации). Их отмечали в виде участков пахигирии (реже диффузной), когда были определены явные признаки упрощенного рисунка мозговых извилин (рис. 4). Реже встречали фокальную микрополигирию.
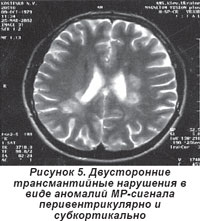
Еще одним частым проявлением фокальной трансмантийной дисплазии у данной категории больных были различные по форме и площади аномалии МР-сигнала, отмечавшиеся вдоль всей мантии головного мозга (рис. 5). Эти изменения трудно трактовать как какой-то определенный порок развития. Возможно, это один из вариантов нарушения архитектоники.
Наряду с пахигирией, вентрикуломегалией, расширением межполушарной борозды у части больных отмечали гетеротопии. Чаще встречали субэпендимальную гетеротопию (узловое скопление нейронов вокруг боковых желудочков), нередко неправильно трактовавшуюся как последствие ишемического поражения головного мозга или как кальцинаты вследствие перенесенной внутриутробной инфекции. У таких больных на разных этапах развития появлялся судорожный синдром. Гетеротопию в виде единичных узелков между корой и желудочками мозга (подкорковая гетеротопия) встречали редко и всегда в сочетании с другими трансмантийными расстройствами. Изолированную гетеротопию без других аномалий мозга у обследованных детей не встречали.
Надо отметить, что у всех больных выявляли различное сочетание различных вариантов дисмиграционных аномалий, что, вероятно, и определяло тяжесть неврологической симптоматики. Опыт показывает, что единичные пороки развития проявляются не такой выраженной неврологической симптоматикой, которая иногда и вовсе отсутствует. При этом единичные аномалии развития, например гетеротопии, определяются как находка. Но угроза появления пароксизмов у ребенка на следующем этапе развития остается.
Полагаем, что в диагностике аномалий головного мозга практическому врачу помогут следующие стандарты.
1. Отсутствие критического периода, в том числе тяжелой гипоксии в неонатальном периоде, при наличии патологии неврологического статуса дает возможность предполагать аномалию развития головного мозга, особенно у доношенного новорожденного.
2. Особое внимание к увеличению размеров желудочков мозга: у недоношенного ребенка изолированная вентрикуломегалия чаще всего является следствием гипоксического поражения нервной системы. У доношенного новорожденного вентрикуломегалия нередко является радиологическим проявлением аномалии мозга.
3. Для дифференциальной диагностики с внутриутробным энцефалитом помогут специфические иммунологические исследования ликвора.
4. При наличии судорожного синдрома в неонатальном периоде ребенок должен пройти радиологическое обследование на наличие аномалий головного мозга.
5. Гипотония в период новорожденности является частым симптомом грубых пороков развития головного мозга.
6. Задержка темпов психомоторного развития и нарушение развития постуральных рефлексов часто также являются синдромами аномалий головного мозга.
Выявление пороков развития головного мозга в как можно более ранние сроки жизни не может быть переоценено. Без своевременной диагностики таких аномалий развития пациент будет обречен получать терапию по поводу гипоксического поражения мозга или внутриутробной инфекции до тех пор, пока диагноз порока мозга не станет очевидным в более старшем возрасте ребенка.
1. Алиханов А.А. Нейрорадиологическая модель различных вариантов нарушения нейронной миграции // Журнал неврологии и психиатрии. — 2004. — №10. — С. 81-85.
2. Бочков Н.П. Вклад генетики в медицину // Журнал неврологии и психиатрии. — 2002. — №2. — С. 3-15.
3. Вельтищев Ю.Е. Актуальные направления научных исследований в педиатрии // Российский вестник перинатологии и педиатрии. — 2003. — №1. — С. 5-11.
4. Вельтищев Ю.Е., Юрьева Э.А. О значении методов лабораторной диагностики для профилактической (превентивной) педиатрии // Российский вестник перинатологии и педиатрии. — 2000. — №5. — С. 6-14.
5. Barkovich A.J., Kuzniecky R.I., Bollen A.W., Grant P.E. Focal transmantle dysplasia: A specific malformation of cortical development // Neurology. — 1997. — V. 49, №4.
6. Cohen M.M., Jr. The Child With Multiple Birth Defects. — Second edition. — New York: Oxford University Press, 1997. — 267 p.
7. Gordon Neil. Epilepsy and Disorders of Neuronal Migration. I Introduction // Developmental Medicine and Child Neurology. — 1996. — V. 38. — Р. 1053-1057.
8. Volpe J. Neurology of newborne. — New York: Raven Press, 1986.