
Журнал «Здоровье ребенка» 2 (45) 2013
Вернуться к номеру
Chronic mucocutaneus candidiasis as a primary immunodeficiency in children
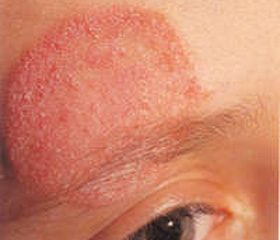
Авторы: Chernyshova L.I.1., Bondarenko A.V. 1, Volokha A.P.1, Lapiy F.I. 1, Chernyshov V.P.2, Marodi L.3, 1 National Medical Academy of Postgraduate Education named P.L.Shupyk, Kiev, Ukraine, 2 SІ “Institute of Pediatrics, Obstetrics and Gynecology”, 3 University of Debrecen, Hungary
Рубрики: Инфекционные заболевания, Педиатрия/Неонатология, Дерматология
Разделы: Справочник специалиста
Версия для печати
Хронічний шкірно-слизовий кандидоз — генетично гетерогенна група захворювань, спричинених мутаціями генів STAT1, IL17RA, IL17F, CARD9, AIRE, STAT3, TYK2, IL12RB1 и IL12B. Хронічний шкірно-слизовий кандидоз може бути синдромом при первинних та вторинних імунодефіцитах і водночас самостійною нозологічною формою первинного імунодефіциту. Основними компонентами імунопатології хронічного шкірно-слизового кандидозу на сьогодні вважають дефекти імунітету, опосередкованого IL-17 та IL-22. У статті наведені два випадки хронічного шкірно-слизового кандидозу у дітей, обумовленого мутаціями гена STAT1.
Хронический кожно-слизистый кандидоз — генетически гетерогенная группа заболеваний, обусловленных мутациями генов STAT1, IL17RA, IL17F, AIRE, STAT3, TYK2, IL12RB1 и IL12B. Хронический кожно-слизистый кандидоз может быть синдромом при первичных и вторичных иммунодефицитах, а также самостоятельной нозологической формой первичного иммунодефицита. Основными компонентами иммунопатологии хронического кожно-слизистого кандидоза на сегодня считаются дефекты иммунитета, опосредованного IL-17 и IL-22. В статье описаны два случая хронического кожно-слизистого кандидоза у детей, обусловленного мутациями гена STAT1.
Chronic mucocutaneous candidiasis is a genetically heterogeneous group of disorders associated with mutations in genes STAT1, IL17RA, IL17F, AIRE, STAT3, TYK2, IL12RB1, and IL12B. Chronic mucocutaneous candidiasis can present as a clinical syndrome in primary and secondary immunodeficiency, or distinct nosological entity of primary immunodeficiency. Today the major components of immunopathology of chronic mucocutaneous candidiasis are defects of immune response, mediated by IL-17 and IL-22. The article describes two cases of chronic mucocutaneous candidiasis, associated with mutations in gene STAT1, in children.
хронічний шкірно-слизовий кандидоз, імунодефіцит, діти.
хронический кожно-слизистый кандидоз, иммунодефицит, дети.
chronic mucocutaneous candidiasis, immunodeficiency, children.
Candida albicans and other Candida species are opportunistic yeasts that are commensal microorganisms of the skin and mucous membranes. Invasive candidiasis appears very seldom, might involve any internal organ or anatomic site and is a significant cause of morbidity and mortality of immunocompromised subjects, particularly those with PIDs affecting granulocytes. Granulocytes, monocytes and macrophages ingesting and killing Candida species yeasts. Phagocytes are of key importance in the host defense against invasive candidiasis [8]. However, mucosal Candida species infections that are self-limited and transient can occur during menstruation and are frequent during pregnancy and in newborn infants.
Chronic mucocutaneous candidiasis (CMC) refers to a heterogeneous group of disorders characterized by recurrent or persistent superficial infections of the skin, mucous membranes, and nails with Candida organisms, usually Candida albicans [11]. Chronic noninvasive candidal infections often associates with autoimmune manifestations (most commonly endocrinopathies).
CMC was first described in 1929 by Thorpe and Handley followed by a more extensive description in the fifties and sixties last century [12].
Chronic mucocutaneous candidiasis is a clinical phenotype of many disorders: immunodeficiencies, endocrinologic, and autoimmune diseases. The unifying feature of these heterogeneous disorders is impaired cell-mediated immunity against Candida species.
1. Chronic mucocutaneous candidiasis as opportunistic infection in primary and secondary combined immunodefiencies.
Persistent and recurrent candidal infections typically occurs as a major disease manifestation in patients with advanced HIV infection, diabetes mellitus, neoplasiae, disorders with prolong use of antibacterial and immunosuppressive agents, and also in patients primary quantitative or qualitative T-cell deficiency: severe combined immunodeficiency, CD25 deficiency, complete DiGeorge syndrome, ataxia-telangiectasia. All these clinical conditions re characterized by general susceptibility to multiple other pathogens and various severe invasive infectious diseases. Besides candidiasis of skin and mucous membranes patients with combined immunodeficiencies have disseminated invasive candidal infections.
2. Chronic mucocutaneous candidiasis as manifestation of infectious syndrome in primary immunodeficiency.
Chronic mucocutaneous candidiasis could manifest as a main clinical syndrome in primary immunodeficiency. CMC is a main feature of infectious syndrome in patients with autoimmune polyendocrinopathy-candidosis-ectodermal dystrophy (autoimmune polyendocrine syndrome type I (APS-I or APS1). It caused by deficiency of AIRE gene (AutoImmune REgulator) and associates with autoantibodies against the cytokines IL-17 (IL-17A and IL-17F) and IL-22 [4, 7]. Cytokines IL-17A, IL17F та IL-22 play crucial role in immunity against Candida. Importantly, anti–IL-17, and anti–IL-22 might serve as serologic markers of the disease in early childhood before manifestations of endocrine organ damage. These antibodies can persist for years without the appearance of antibodies to endocrine organs or any clinical manifestation of APS-1 [6].
Autoimmune polyendocrinopathy-candidosis-ectodermal dystrophy is known for its wide variation in clinical presentation and course of disease, even among affected family members carrying an identical genetic aberration. In addition, the disease may present with rare manifestations that persist for many years [11]. The first manifestation of the disease can start between the ages of 2 months to 18 years. The classic triad is mucocutaneous candidiasis, hypoparathyroidism, and adrenal failure. Chronic, or sometimes recurrent, candidal infection of the oral cavity, nails, and skin, and less frequently the esophagus, vagina, and gastrointestinal tract, is the presenting feature in 60 percent of patients. Hypoparathyroidism is the most common endocrine abnormality in this disease and the second most common feature in AIRE deficiency, occurring at presentation in about 30 percent of patients. The resultant hypocalcemia and hypomagnesemia is sometimes hard to control and may lead to seizures. Adrenal failure is the third most common feature in the disease. It occurs in more than 60 percent of cases by the age of 15 years. Other endocrinopathies include type 1 diabetes mellitus, hypothyroidism, growth hormone deficiency, ovarian failure (appears to coexist with adrenal failure), and male hypogonadism. Other autoimmune manifestations include vitiligo and alopecia areata, which affects more than 30 percent of patients after the age of 20 years
The patients with autosomal dominant hyper-IgE syndrome can have clinical phenotype with candidal infections of skin and mucous membranes [6]. The immunodeficiency caused by dominant-negative mutations of STAT3, which lead to severe depletion of CD4+ T cells and decreased production of IL-17A and IL-22. These patients have very small number of Th17 as result diminished immune response to IL-6, IL-21 and IL-23. Chronic mucocutaneous candidiasis associates with severe staphylococcal infections of lung and skin. New autosomal recessive variant of the hyper-IgE syndrome recently described in Japan in patient with a homozygous mutation in TYK2. The patient presented with atopic dermatitis, markedly increased serumIgE titers, and recurrent infections with Staphylococcus species, herpes simplex virus (HSV), BCG, and Candida species. The patients with TYK2 deficiency has decreased immune response of T lymphocytes to stimulation by IL-6 and differentiation of CD4+ Т- lymphocytes Th17.
Patients with autosomal recessive IL-12p40 or IL-12Rβ1 deficiency suffer from Mendelian susceptibility to mycobacterial disease (MSMD) and occasionally develop mild chronic mucocutaneous candidiasis [4, 12]. Recurrent oropharyngeal candidiasis has been found in 24% of 141 reported patients with IL-12Rβ1 deficiency. Chronic mucocutaneous candidiasis has also been observed in 7.5% of 44 patients with IL-12p40 deficiency. T cells from patients with IL12RB1 mutations cannot respond to either IL-12 or IL-23 because these cytokines bind IL-12Rβ1. Patients with IL-12p40 deficiency, a subunit shared by IL-12 and IL-23, might be prone to chronic mucocutaneous candidiasis because of impaired IL-23–dependent immunity, which is important for the maintenance of IL-17–producing T lymphocytes.
3. Chronic mucocutaneous candidiasis as separate nosological form of primary immunodeficiency
Several new forms of chronic mucocutaneous candidiasis as separate disease entities of primary immunodeficiency were described in the last years [2,4,9, 10]. These disorders characterized impairment of IL-17 production or absence IL-17 receptor. Regarding last WHO/IUIS classification of primary immunodeficiency (2011) chronic mucocutaneous candidiasis is caused by mutations of IL17RA, IL17F, CARD9, STAT1 genes and refers to deficiencies of innate immunity [1].
In 2011 group of immunologists reported two genetic etiologies of chronic mucocutaneous candidiasis: autosomal recessive deficiency in the cytokine receptor, interleukin-17 receptor A (IL-17RA), and autosomal dominant deficiency of the cytokine IL-17F. Congenital impairment of immune response, mediated by cytokines IL-17A, IL17F and IL-22, results in recurrent infections, caused by Candida albicans and, to a lesser extent, Staphylococcus aureus. In a Moroccan patient born to consanguineous parents, an autosomal recessive mutation of the IL17RA gene, leading to complete loss of protein expression, was found to cause chronic mucocutaneous candidiasis with neonatal onset [10]. He presented with C. albicans dermatitis during the neonatal period and displayed S. aureus dermatitis at five months of age.
In 2011 were described first 12 cases of autosomal dominant form of chronic mucocutaneous candidiasis, caused by a monoallelic gain-of-function mutation in the coiled–coil (CC) domain of signal transducer and activator of transcription 1 (STAT1) [5]. Previously described heterozygous STAT1 mutant alleles are loss-of-function and cause autosomal dominant predisposition to mycobacterial disease caused by impaired STAT1-dependent cellular responses to IFN-γ. Other loss-of-function STAT1 alleles cause autosomal recesive predisposition to intracellular bacterial and viral diseases, caused by impaired STAT1-dependent responses to IFN-α/β, IFN-γ, and IL-27.
Stronger cellular responses to the STAT1-dependent IL-17 inhibitors IFN-α/β, IFN-γ, and IL-27, and stronger STAT1 activation in response to the STAT3-dependent IL-17 inducers IL-6 and IL-21, hinder the development of T cells producing IL-17A, IL-17F, and IL-22. This results in impaired IL-17 T-cell development and IL-17 T cell–mediated antifungal responses. One of 12 described patients with autosomal dominant chronic mucocutaneous candidiasis caused by gain-of-function STAT1 alleles observe by authors during 13 years (case 1).
Recurrent fungal infection typical for the patients with autosomal recessive CARD9 deficiency, firstly reported in 2009 in a large, consanguineous, 5-generation Iranian family [3]. Caspase recruitment domain (CARD) 9 deficiency predispose 7 members of family to candidal disease. Only 4 of the 7 patients had chronic mucocutaneous candidiasis since early childhood, and 1 patient presented with invasive candidiasis without superficial form. Three members of family died during adolescence after invasive infection of the brain with Candida species. In all 7 patients and their 18 relatives were identified homozygous mutations of CARD9 gene. The clinical picture of chronic mucocutaneous candidiasis is less severe than in autosomal dominant CMC. Imunological investigation of family members with candidal infections revealed significant decrease of Th17 (0,2% CD4+CD45RO+IL-17+IFN-γ-cells compared to1,2% Th17 in control)
Other genetic aberrations, as Lyp and dectin-1 mutations, associated with features of chronic mucocutaneous candidiasis were found in several families [12].
Lymphoid phosphatase (Lyp) interacts with the Src family negative regulatory kinase and downregulates TCR signaling. Lyp deficiency enhances T cell activation resulting in possible compromised central as well as peripheral tolerance. All patients with chronic mucocutaneous candidiasis who carried a mutation of PTPN22, which encodes phosphatase Lyp had oral or skin candidiasis, similar to patients with AIRE mutations. The most common endocrine abnormalities were hypothyroidism and gonadal failure.y. A unique finding seen in five of the patients with the Lyp mutant was chronic lung disease including bronchiectasis.
Candida albicans is recognized by the innate immune system through pattern-recognition receptors, such as the toll-like-receptors (TLRs) and lectin like-receptors (Dectin-1 and Dectin-2). TLR2, TLR4, and mannose receptors recognize mannans, Candida cell wall components. These receptors collaborate with the beta-glucan receptor Dectin-1 in stimulation of cytokine production. Dectin-1 enhances TLR2 and TLR4 induced cytokine production [12]. The patients with Dectin-1 deficiency have non-invasive chronic candidal infections of skin and mucous membranes [4].
The scientists now are only in the beginning to elucidate the molecular genetic mechanisms and phenotypic characteristics of most forms of chronic mucocutaneous candidiasis. Cases with abnormal IL-17 immunity (through impaired IL-17 or IL-17 receptor function or STAT1 gain-of-function mutation) serve as naturally occurring experiments, the results of which support the hypothesis that CMC can be caused by deficiencies. IL-17F and IL-17RA deficiencies and gain-of function mutations of STAT1 alleles have been described with only CMC (CMCD).
However, it is important to emphasize that in some of these conditions, genotype-phenotype correlations are difficult to establish because of the limited number of patients described (eg, IL-17 deficiency, IL-17 receptor deficiency, TYK2 deficiency, and CARD9 deficiency).
Clinical manifestations. Patients present with recurrent or persistent superficial candidal infections of the oral cavity (thrush), skin and nails, caused by mostly Candida species
Cutaneous dermatophyte infections also common. Candidal infections have recurrent refractory features. However, disseminated forms of Candida infections occur very seldom, due to intact function of neutrophiles. The patients with chronic mucocutaneous candidiasis can have enamels dysplasia, malabsorbtion, keratoconjunctivitis, nail dystrophy.
Chronic mucocutaneous candidiasis often associated with autoimmune endocrinopathy. Autoimmune manifestations other than endocrinopathy, including autoimmune hemolytic anemia, immune thrombocytopenia purpura, autoimmune neutropenia, autoimmune hepatitis and vitiligo have been reported.
Susceptibility to bacterial and viral infections was identified in many patients with]. Severe lung infections with bronchiectasis described in patients with hypogammaglobulinemia, sometimes even with normal level of immunoglobulines but impared response to polysaccharide antigens.
Diagnosis. The diagnosis of candidiasis is confirmed by observation of mycelial forms on microscopic examination. Since Candida yeasts (especially C. albicans) are normal inhabitants of the skin and oral mucosa, it must always be noted that positive culture does not always indicate the presence of candidal infection.
The diagnosis of chronic mucocutaneous candidiasis is suspected in patients with typical clinical picture. All patients with chronic or recurrent candidal infections should be evaluated for primary and secondary immunodeficiencies. Consider a complete blood cell count, to screen for leukopenia, an HIV test, immunoglobulin levels including IgE level, and B and T cell subpopulations.
Immune studies in many patients do not revealed any changes of number of lymphocytes subsets. However, some patients have lymphopenia and reduced number and/or function of circulating T cells. Evaluation of the immune system may identify a selective inability to respond in vitro (T cell proliferation) or in vivo (cutaneous delayed-type hypersensitivity) to Candida. In others, more extensive abnormalities in vitro antigenic and mitogenic responses are identified .[11, 12]. Investigation of lymphosytes subsets can found in some patients decreased number of CD4+IL-17+-producing Т-cells (Th17) and production IL-17 and IL-22. Molecular diagnostic is important for verification of nosologic form of primary immunodeficiency with features of chronic mucocutaneous candidiasis.
Screening laboratory tests for endocrine dysfunction, associated with chronic mucocutaneous candidiasis include blood glucose or glycosylated hemoglobin testing, thyroid function tests, liver functions tests, serum electrolyte evaluation, corticotropin testing, and serum cortisol values. Other endocrine screening tests that may be considered include follicle-stimulating hormone, luteinizing hormone, prolactin, testosterone, parathyroid-stimulating hormone, calcium, phosphate, magnesium. Baseline and yearly follow-up tests should be performed to screen for associated endocrinopathy.
Humoral immunity may also be affected, including low IgG2 and IgG4, IgA and IgM deficiency, hypogammaglobulinemia, and an inadequate response to vaccination with polysaccharide antigens (eg, unconjugated pneumococcal vaccine).
Treatment. Management of chronic mucocutaneous candidiasis can be difficult, and relapse is common following discontinuation of therapy. Topical therapies are not usually effective in these patients.
Systemic antifungal therapy is the mainstay of therapy of chronic mucocutaneous candidiasis. Management includes also treatment of associated endocrine and autoimmune abnormalities and other infections of viral and bacterial etiology.. In case of ineffective aggressive courses of antifungal medications consider prolonged repeated courses every 3-4 months with 4-6 weeks duration or chronic suppressive therapy. Fluconazole is the preferred treatment.
Drug resistance may occur to fluconazol, another azole agent will need to be used: itraconazole, voriconazole, or posaconazole. Liver function should be carefully monitored while patients are on systemic therapy with these drugs. Amphotericin and echinocandines has been successfully used in severe cases.
Antibody deficiency with severe manifestations should be treated with immune globulin replacement.
Case 1. Chronic mucocutaneous candidiasis (gain-of-function mutation in the CC-domain of STAT-1 gene)
A 20-years old man is under our observation since 7 years. He is suffering from recurrent oral candidiasis since 3.5 months of age. Two pneumonias were observed up to 12 months, since 12 months persistent cough was present. The negative results of HIV testing and negative sweat chloride test allowed to exclude HIV infection and cystic fibrosis. At the age of 17 months onychomycosis, frequent recurrences of oral candidiasis. Mycological study highlighted C. albicans. At 5 years microspores were diagnosed. Good therapeutic effect in treating with grizeofunvin was achieved. Since 5 years of life candidiasis of the skin and mucous membranes became continuous-recurrent with a temporary response to antifungal drugs and the gradual development of resistance.
/!eng/8/8.jpg)
At the age of 8 boy had a severe pneumonia, caused by H. infuenzae type b, accompanied by purulent endobronchites. The child could not attend school. Significant decline in height and body weight (under 25 centiles), subtotal alopecia and autoimmune keratouveitis were revealed at his first admission to our center. Endoscopy demonstrated candidal esophagitis and esophageal stenosis. Immunologic testing revealed lymphopenia, decreased number of T-, B- and NK lymphocytes.
During the study of the function of lymphocytes moderate reduction in reaction blast transformation with mitogen (fitohemahlyutynin, konkonavalin), a significant reduction in the proliferative response to core antigens (tetanus toxoid, tuberculin), almost complete lack of response to antigens of infectious agents - viruses (herpes simplex virus, influenza B) and Candida antigen were observed (Table 2).
/!eng/8/8_2.jpg)
Decreased IgG level was also detected during immunological investigation. (Table .3). IgE levels within the normal range 45 IU / ml.
/!eng/8/8_3.jpg)
Сomplete absence of IL-17A production by patient's cells was found in response to stimulation with Candida in the study of IL-17A and IL-22 cytokine production in vitro (Table 4).
/!eng/8/8_4.jpg)
Chronic mucocutaneous candidiasis was diagnosed on the basis of characteristic clinical features and results of immunological studies.
Conducted genetic testing at the age of 18 excluded APECED and CARD9. The systematic study of genome revealed heterozygote gain-of-function mutation in СС-domaine of STAT1-gene (D165G), confirming the diagnosis of chronic mucocutaneous candidiasis with an autosomal dominant mode of inheritance as an independent nosological form of primary immunodeficiency. Mutation is absent in parents and younger brother, therefore, occurred in the patient de novo.
The main methods of treatment are antifungal and antibacterial treatment of infectious episodes and prophylactics. Due to the presence of purulent foci of infection (chronic suppurative endobronchites, bronchiectasis, chronic haymoroetmoyidyt) together with moderate reduction in serum immunoglobulin IgG (Table 3) at age of 13 it was decided to start a permanent monthly intravenous immunoglobulin replacement therapy at a dose of 400 mg/kg per month, which made a positive impact on the disease. But replacement therapy is not irregularly because of the high cost of treatment. At age of 14, severe destructive pneumonia developed.
Because of chronic skin and mucous candidiasis, the boy developed total alopecia, formed a double esophageal stenosis. Continuous recurrence of respiratory infections such as purulent bronchitis led to the bronchiectasis formation with accompanying signs of chronic hypoxia
The patient has significant growth and weight retardation (22 kg at the age of 18 years old). The concentration of adrenocorticotropic hormone, cortisol, parathyroid hormone, free thyroxine, testosterone and insulin-like growth factor-1 are in the normal ranges. The patient’s condition is serious due to severe heart and respiratory failure.
Case 2. Chronic mucocutaneous candidiasis (mutation of the DNA-binding domain of STAT-1).
The patient O., born in 1999, is under our supervision since 5 years old. The child was born from pregnancy ІІІ (previous 2 - miscarriage), labor I. Weight at birth 4000 g. Umbilical wound had healed at the age of 1,5 months.
Since birth the child had recurrent oral candidiasis and cheilitis, since 3 months – dermatitis. Microbiological cultures repeatedly yielded C.albicans from buccal and gingival swabs. At the age of 8 months underwent widespread thrush in the mouth with further relapses for every 3-4 weeks. Aphthae associated with marked pain syndrome. Aphthae represent as wounds from 0.5 to 1 cm in diameter, covered with yellow-white accretions healing of up to 10-14 days, occurring mainly on the tongue and inner surface of the lips. Deterioration in the oral cavity is often accompanied by diarrhea that responds to antifungal drugs. Nystatin, fluconazole, itraconazol are mostly used as a treatment.
He first presented with onychomycosis at 8 months of age which eventually was cured by long-term use of itraconazole (about one year), and candidal paronychia at 2½ years of age.
At the age of 5 years because of complains of retrosternal pain on swallowing solid food the esophageal narrowing of a size of 0,4 cm in diameter and 4 cm length in the middle third of the esophagus was detected by chest radiography with using contrast media. At gastroduodenal fibroscopy down to 18 cm from the incisor teeth the mucosa of the esophagus showed multiple erosions with fibrin and a stricture of about 35 mm long. The esophageal stricture developed probably due to the recurrent esophagitis. Several air dilatations of the esophagus were performed without significant improvement. He underwent plastic esophageal surgery at age 6 because of persistent vomiting.
At the age of 8 years underwent surgery for phimosis, which developed as a result of recurrent balanoposthitis.
He had recurrent bronchitis and several episodes of bacterial pneumonia which resolved after parenteral administration of antibiotics.
He has suffered from herpes labialis rash on the lips since 3 years old with recurrences of 6-10 times per year.
The patient also had widespread tinea (10 years) as an annular rash 10-15 cm in diameter on the skin of the trunk, arms and legs. Trychophyton were identified from skin lesions. Lesion was stopped by system using of griseofulvin.
Recurrent X-ray-proven bilateral sinusitis is observed since 12 years with recurrences to 6 times per year improving on oral β-lactam antibiotics, most of the time amoxicillin-clavulanic acid. Str. pyogenes, S.epidermidis, S. аureus were detected in nasopharyngeal swabs.
HIV infection and cystic fibrosis were excluded. Antibodies to HSV1/2 are negative. Family history is not burdened. Patient has a younger sister, healthy.
Common blood did not reveal any pathological changes. Serum IgG and IgM are within normal range, IgA is moderately increased (Table 5.). IgE levels are also normal (30 IU/ml). Lymphocyte subpopulations at the age of 5 were normal (Table 6) with no changes with age.
/!eng/8/8_5.jpg)
Positive antibodies to dsDNA were detected at the age of 9 (4,3 unites (N<1,1)) and 11 years old (1,2 unites (N <1,1)). Antiphospholipid antibodies were also positive: aCL IgG/IgM 14 GPL (norma <10 GLP), aPS gG/IgM – 40 U/ml (norma <10 U/ml), aPE IgG/IgM – 40 U/ml (norma <12 U/ml) at the age of 9, aCL IgG/IgM 20 GPL (norma <10 GLP), aPS gG/IgM – 30 U/ml (norma <10 U/ml), aPE IgG/IgM – 45 U/ml (norma <12 U/ml) at the age of 11. Antinuclear antibodies are negative.
/!eng/8/8_6.jpg)
Based on clinical data (recurrent oropharyngeal candidiasis, Candida esophagitis, onychomycosis) the presumptive diagnosis of chronic mucocutaneous candidiasis was established.
Because chronic mucocutaneous candidiasis is one of the manifestations of APECED (autoimmune polyglandular syndrome type I) together with the fact of positive autoantibodies at the age of 10 years molecular genetic study was carried out (Department of Pediatric Infectious Diseases and Clinical Immunology, University of Debrecen) regarding the diagnosis of APECED. However AIRE gene mutations have not been identified.
Because of the presence of resistant recurrent mucocutaneous candidiasis and exclusion of other known immunodeficiency, further diagnostic molecular genetic search was carried. As a result the T385M mutation in the DNA-binding domain of STAT1 gene was detected.
Today the boy suffers from chronic bronchitis with exacerbations 3-4 times a year, has chronic sinusitis, recurrent aphthous stomatitis. In order to prevent a recurrence of chronic mucocutaneous candidiasis receives courses of fluconazol, ketoconazol, itraconazol with unsustainable effect, resistance developed rapidly, the constant need to change drugs.
When determining specific antibodies to vaccine antigens a reduction in production of specific antibodies to protein and polysaccharide antigens was demonstrated: anti-Str. pneumoniae (mg/ml) 0.38 (rate>10.00), anti-H. influenzae type B (mg/ml) 1.98 (normal> 0.15), anti-tetanus (IU/ml), 0.08 (normal>0.70). Take into account recurrent and chronic bacterial infections and impaired antibody production to specific antigens, the replacement therapy with intravenous immunoglobulin was started.
CONCLUSION.
- Chronic mucocutaneous candidiasis can be one of features of infectious syndrome in patients with primary and secondary immunodeficiencies, but can exist as a separate nosologic form of primary immunodeficiency..
- The first time in Ukraine we described clinical cases of chronic mucocutaneous candidiasis as a separate variant of primary immunodeficiency in two boys with mutation of STAT1 gene and impairment of IL-17-mediated immune response. These clinical cases confirmed the hypotheses that chronic mucocutaneous candidiasis can be caused by defect of defense mechanisms on body surfaces, mediated by cytokines with key role of IL-17.
- We found different types of mutations of STAT1 gene (gain-of-function mutation in СС-domain and mutation of DNA-binding domain) in observed patients with similar clinical phenotype of chronic mucocutaneous candidiasis. Mutation of DNA-binding domain of STAT1 gene reported now only in several patients with chronic mucocutaneous candidiasis.
- The chronic candidal infections of skin, nails and mucous membranes associated in our patients with chronic and recurrent bacterial (bronchitis, pneumonia and sinusitis) and viral infections (especially herpes viruses).
- Al-Herz W., Bousfiha A., Casanova J-L., et al. Primary immunodeficiency diseases: an update on the classification from the International Unit of Immunological Societies Expert Committee for Primary Immunodeficiency// Frontiers in Immunology 2011. – V2, N54.- P.1-26.
- Gaffen S.L. Recent advances in the IL-17 cytokine family//Current Opinion Immunology 2011.- V.23, N5.- P.613-619.
- Glocker E-O., Hennigs A., Nabavi M., et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections//N Engl J Med 2009. – V.361.- P.1727-35.
- Hanna S., Etzioni A. New host defencse mechanisms against Candida species clarify the basis of clinical phenotypes//J Allergy Clin Immunology 2011. – V.127. – P.1433-7
- Liu L., Okada S., Kong X-F., et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneus candidiasis//J Experimental Medicine 2011. – V. 208, N8.- P.1635-1648.
- Marodi L., Cypowyj S., Toth B., Chernyshova L., et al. Molecular mechanisms of mucocutaneus immunity against Candida and Staphylococcus species//J Allergy Clin Immunology 2012. – V.130. – P.1019-27
- Mazza C., Buzi F, Ortolani F. et al. Clinical heterogeneity and diagnostic delay of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome // Clin Immunol 2011. – V. 139. – P. 6-11.
- Netea M.G, Marodi L. Innate immune mechanisms for recognition and uptake of Candida species //Trends Immunology, 2010. – V.31. – P. 346-353.
- Ochs H.D, Oukka M., Torgerson T.R. Th17 cells and regulatory T cells in primary immunodeficiency diseases//J Allergy Clin Immunology 2009. – V.123. – P.977-83
- Puel A., Cypowyj S., Bustamante J., et al. Chronic mucocutaneus candidiasis in humans with inborn errors of interleukin-17 immunity//Science 2011.- V.332, N 6025. – P. 65-68.
- Robles D.T. Chronic mucocutaneus candidiasis // www.emedicine.medscape.com
- Roifman CM. Chronic mucocutaneus candidiasis //www.uptodate.com
- Van de Veerdonk F.L., Theo S. Plantinga T.S., Hoischen A. STAT1 Mutations in autosomal dominant chronic mucocutaneous candidiasis //N Engl J Med 2011. – V.365.- P.54-61.