Статья опубликована на с. 118-122
Генодерматозы являются наследственными заболеваниями кожи, включающими порядка сотни нозологических форм, проявляющихся нарушением ороговения и пигментации, дистрофическими изменениями кожи и ее придатков, невоидными и опухолевыми процессами. Эта проблема требует детального изучения и решения вопроса реабилитации больных. Данная группа дерматозов довольно часто сочетается с болезнями нервной системы (факоматозы), эндокринной, костной и др. Одним из таких заболеваний является ихтиоз, представляющий собой наследственное заболевание кожи, характеризующееся диффузным нарушением ороговения по типу гиперкератоза с образованием на коже чешуек, напоминающих рыбью чешую. Частота встречаемости патологии составляет 1 : 3000 — 1 : 4500 населения [3, 7, 9].
Несмотря на то что врожденные ихтиозы являются относительно редкими заболеваниями как в педиатрической, так и в дерматологической практике, их актуальность высока и обусловлена тяжелым клиническим течением, нередко приводящим к инвалидизации пациентов, сложностью распознания и дифференциальной диагностики, резистентностью к проводимой терапии, необходимостью постоянного медицинского контроля [1, 3]. Следовательно, накопленные знания по данной нозологии позволят своевременно установить клинический диагноз и назначить лечение, которое сможет не только спасти жизнь пациентов, но и улучшить ее качественную составляющую.
Выделяют следующие формы ихтиоза: вульгарную (аутосомно-доминантный тип наследования), Х-сцепленную рецессивную (самостоятельная форма), ихтиоз плода, врожденную ихтиозиформную эритродермию (ламеллярный и эпидермолитический ихтиоз), одностороннюю, линеарную огибающую, иглистую. А также ихтиоз может дополнять в качестве одного из симптомов такие наследственные синдромы, как синдром Нетертона, Рефсума, Руда, Шегрена — Ларссона, Гоше 2-го типа и другие [6]. Указанные наследственные синдромы клинически проявляются не только ихтиозом, но и поражением иных внутренних органов и систем, в частности нервной системы.
Так, известный синдром Шегрена — Ларссона сопровождается ихтиозом и такими неврологическими нарушениями, как спастические параличи, олигофрения, дегенеративный ретинит, амблиопия. Синдром Рефсума характеризуется сочетанием ихтиоза с гипертрофической полинейропатией, мозжечковой атаксией, глухотой, атипичным пигментным ретинитом и изменениями на ЭКГ. Более полиморфная клиническая картина представлена при синдроме Гоше 2-го типа. Наряду с ихтиозом у пациентов отмечается выраженная неврологическая симптоматика в виде глазодвигательных расстройств (страбизм), спастических параличей с контрактурой суставов.
Первые признаки обычного ихтиоза в виде сухости и шелушения проявляются на третьем месяце жизни или несколько позже (в возрасте первых 2–3 лет жизни). Основные клинические симптомы заболевания развиваются постепенно, достигая максимума к периоду полового созревания. Помимо специфических кожных изменений у таких пациентов отмечаются: функциональная недостаточность эндокринной системы (щитовидной, половых желез), снижение активности В- и Т-клеточного иммунитета, склонность к аллергическим заболеваниям, низкая сопротивляемость к пиококковым и вирусным инфекциям и др. [10].
При всех формах ихтиоза страдают процессы терминальной клеточной дифференцировки и ороговения эпидермиса, что обусловлено мутациями либо нарушением экспрессии генов, кодирующих различные типы кератина [3], а также другие маркеры дифференцировки: структурные протеины клеточной оболочки (лорикрин, инволюкрин), промежуточный филаментассоциированный протеин — профилаггрин, ферменты, участвующие в кератинизации, — трансглутаминаза. Возможно вовлечение разноименных генов, что объясняет широту спектра клинических проявлений заболевания [1–5, 7, 8].
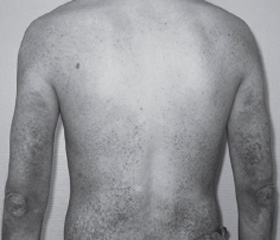
Клиническая картина вульгарного ихтиоза характеризуется диффузным поражением кожных покровов тела с наибольшим вовлечением в процесс кожи разгибательных поверхностей конечностей (коленные и локтевые суставы) при отсутствии подобных изменений на сгибательных поверхностях тех же коленных и локтевых суставов, кожи в подмышечных впадинах и шеи (рис. 1). Специфика данных кожных проявлений заключается в различной степени выраженности наслоения чешуек разного размера и цвета — от белого до черного, благодаря чему кожа становится сухой и шершавой на ощупь. В детском возрасте вовлечение в процесс кожи лица встречается редко, тогда как у взрослых может наблюдаться шелушение лба и щек. Часто отмечается поражение придатков кожи. Так, ногтевые пластины становятся сухими и ломкими, шероховатыми, деформированными, волосы имеют тенденцию к истончению и разрежению (рис. 2).
/119-1.jpg)
/119-2.jpg)
Х-сцепленный ихтиоз был выделен из вульгарного ихтиоза на основании гистологических и генетических исследований. Основным дефектом является дефицит стеролсульфатазы и арилсульфатазы, приводящий к отложению в эпидермисе избытка холестерина сульфата, ретенционному гиперкератозу и нарушению процессов десквамации. При гистологическом исследовании в отличие от вульгарного ихтиоза зернистый слой утолщен, а не истончен [10, 12].
Ихтиоз плода (плод Арлекина) представляет собой особую форму ихтиоза, несовместимую с жизнью. Ребенок, как правило, рождается преждевременно и живет не более нескольких недель. Заболевание имеет аутосомно-рецессивный тип наследования, обусловленный мутацией в гене ABCA12. Проявляется образованием толстого панциря из многочисленных чешуек с глубокими трещинами по всей поверхности кожного покрова и сочетается с недоразвитием внутренних органов. Такие новорожденные чаще всего погибают от вторичной инфекции, недостаточности питания, тяжелой анемии, нарушения кровообращения или почечной недостаточности [10, 12].
Врожденная ихтиозиформная эритродермия — форма врожденного ихтиоза, отличающаяся от вульгарного ихтиоза наличием воспаления с рождения. Различают два ее типа — небуллезный и буллезный. Небуллезная врожденная ихтиозиформная эритродермия передается по аутосомно-рецессивному типу наследования, проявляется нарушением ороговения кожи, сопровождающимся сильным шелушением и эритродермией без формирования пузырей при рождении [2, 6, 11].
Буллезная врожденная ихтиозиформная эритродермия наследуется по аутосомно-доминантному типу и проявляется эритродермией, протекающей с образованием пузырей, наличием симптома Никольского, гиперкератотическими линейными очагами. Кожа ладоней и подошв утолщена, беловатого цвета, эктропиона нет. Исходя из того, что у новорожденного на фоне эритродермии возможно наличие большого количества пузырей и эрозий, данное состояние иногда ошибочно расценивается как буллезный эпидермолиз или синдром стафилококковой ошпаренной кожи. По окончании периода новорожденности образование пузырей прекращается или они становятся единичными, а генерализованный гиперкератоз усиливается [3, 9, 12].
Таким образом, клиническая картина ихтиоза преду-
сматривает диффузное поражение кожных покровов, которое нередко сочетается с полиорганной недостаточностью и развитием неврологических нарушений со стороны как центральной, так и периферической нервной системы.
Патофизиологические механизмы указанных
осложнений сложны. Они, вероятно, связаны с нарушением вегетативной, нервно-гуморальной регуляции, гормональными, метаболическими изменениями в организме, связанными с аномальным синтезом кератина.
Для иллюстрации особенностей клинического течения ихтиоза приводим собственные клинические наблюдения.
Больной Ш., 1978 года рождения, инвалид третьей группы. При обращении к дерматологу (дата 12.07.12) жаловался на покраснение кожи, сухость, шелушение, стягивание кожи, слабость в ногах, больше в левой, боли в спине по ходу грудного и поясничного отделов позвоночника, усиливающиеся при ходьбе и при нагрузках, болезненность первого пальца левой стопы, онемение нижней половины тела, затрудненное мочеиспускание.
Из анамнеза заболевания: поражение кожи обнаружено при рождении (1978), установлен диагноз «врожденная ихтиозиформная эритродермия Брока». В семье подобное заболевание отмечается у отца, прабабушки по отцовской линии, родной сестры (инвалид детства).
В возрасте 12–13 лет начали беспокоить боли в мелких суставах кистей с последующей их деформацией и нарушением функции. С 15 лет периодически стали беспокоить боли в позвоночнике давящего и периодически стреляющего характера, сопровождающиеся потемнением в глазах, затруднением ходьбы. В этом же возрасте случайно выявлена протеинурия, появилось затрудненное мочеиспускание.
Находился на диспансерном наблюдении по месту жительства по поводу ихтиоза. Неоднократно получал амбулаторное и стационарное лечение. Периоды обострений сменялись периодами улучшений, однако отмечалось постепенное прогрессирование болезни как со стороны кожи, так и других органов.
Результаты неврологического осмотра. Астенизирован. Сосредоточен на своих ощущениях и заболевании. Со стороны черепных нервов без особенностей. Сухожильные и периостальные рефлексы: с рук D = S, живые; коленные — D > S, оживленные; ахилловы — D > S, высокие. Поверхностные брюшные рефлексы снижены, глубокие брюшные рефлексы оживлены. Стопных и кистевых патологических рефлексов нет. Амиотрофий нет. Сила мышц — 5 баллов в руках, 3,5 балла в ногах. Походка паретична, ходит с тростью. Проба Ромберга отрицательная. Атаксии в конечностях нет. Гипалгезия по проводниковому типу с уровня Th7 симметрично. Снижено суставно-мышечное чувство, больше в левой стопе. Тазовые функции нарушены — задержка мочи.
У пациента имеют место следующие неврологические синдромы: умеренный спастический нижний парапарез, снижение функции тазовых органов, проводниковый тип сенсорных расстройств с уровня Д7-Д8 спинальных сегментов, радикулярный синдром L3-L4, L5-S1. Неврологическая симптоматика обусловлена поражением боковых, частично задних канатиков спинного мозга, задних корешков L5-S1, сегментов спинного мозга.
Характер имеющегося патологического процесса — сосудистый, обусловленный васкулитом сосудов спинного мозга, и вертеброгенный, в связи с выраженным дегенеративным процессом в поясничных межпозвоночных дисках.
Status localis — кожный процесс носит универсальный характер. Кожа гиперемирована, умеренно инфильтрирована. Выражена сухость. Кожа покрыта серо-бурыми чешуйками, плотно сидящими. В области суставных поверхностей — роговые наслоения, в области естественных складок — эритема, гиперкератоз. Ладонно-подошвенный гиперкератоз и гипергидроз. Ногтевые пластинки изменены в цвете, деформированы. Волосы, брови и ресницы в норме. Кожа лица розовая, сухая. Суставы кистей деформированы.
Обследование: RW крови отрицательное. Нb — 149 г/л, Эр 4,7 • 1012/л, Ц.П — 0,9, Л — 4,9 • 109/л, п — 5, с — 53, э — 2, л — 33, м — 7, СОЭ — 6 мм/ч. Мочевина — 14,0; билирубин (общ.) — 7; АлАТ — 0,13; тимоловая проба — 3,1; глюкоза — 4,0; общий белок — 84; альбумины — 60; глобулины — 40; общий анализ мочи без патологии. ФГ ОГК — без патологии. Электронейромиография: имеются признаки нарушения проведения импульса в корешковых отделах L5-S1 слева. Нельзя исключить заинтересованность пирамидного пути с двух сторон. Магнитно-резонансная томография грудного отдела позвоночника: признаки сколиоза, спондилеза, на уровне D8-D9 в спинном мозге визуализируются мелкие единичные очаги с четкими контурами. Магнитно-резонансная томография пояснично-крестцового отдела позвоночника: начальные проявления остеохондроза пояснично-крестцового отдела с протрузиями дисков L3-L4, L5-S1. Магнитно-резонансная томография головного мозга: очаговых изменений не выявлено.
Консультирован неврологом: врожденная ихтиозиформная эритродермия Брока, осложненная системным васкулитом с поражением сосудов почек и спинного мозга, вторичный миелит с уровнем поражения сегментов Д7-Д8, умеренно выраженный спастический нижний парапарез с нарушением статико-локомоторных функций, тазовые нарушения (эпизоды затрудненного мочеиспускания, императивные позывы). Остеохондроз позвоночника, преимущественно поясничного отдела, с протрузиями дисков L3-L4, L5-S1 (по данным МРТ), рефлекторный болевой синдром.
Офтальмолог: диск зрительного нерва бледно-розовый, границы четкие, сосуды в ходе и калибре не изменены.
Ревматолог: системный васкулит с поражением сосудов почек; хронический пиелонефрит, мочевой синдром, вторичная нефропатия.
Травматолог: остеохондроз с преимущественным поражением поясничного и нижнегрудного отделов позвоночника, стойкий болевой синдром, выраженное нарушение опорной функции и движения. Начальные проявления полиостеоартроза с преимущественным поражением мелких суставов кистей и крупных суставов левой нижней конечности.
В данном клиническом наблюдении ихтиоз сопровождается поражением почек, мелких и крупных суставов конечностей, позвоночника, спинного мозга, преимущественно боковых и задних канатиков, системной васкулопатией (васкулитом). Это характеризует ихтиоз как заболевание, инициирующее системное поражение кожи и, возможно, иных компонентов соединительной ткани, структурных протеинов клеточных оболочек, сосудов кожи, внутренних органов, центральной нервной системы.
Больная С., 1982 г. рождения, инвалид детства, родная сестра больного Ш. При обращении к дерматологу жаловалась на сухость и поражение кожи с вовлечением ладоней и подошв, изменения ногтевых пластинок, боли в области сердца, позвоночника, особенно области онемения в руках и ногах, поясницы, головные боли, больше слева, снижение зрения на левый глаз, множественный кариес.
Из анамнеза: болеет с рождения. Установлен диагноз «врожденная буллезная ихтиозиформная эритродермия». Наследственность отягощена: отец, прабабушка по отцовской линии, родной брат имеют такое же заболевание. Наблюдается по месту жительства.
В возрасте 9–10 лет появились периодические боли в области сердца. С 16-летнего возраста присоединились регулярные головные боли. Улучшения наступают после сеансов массажа воротниковой зоны, который проводит регулярно. В этом же возрасте стала отмечать снижение остроты зрения на левый глаз. В 17-летнем возрасте, как и у брата, возникли проблемы с позвоночником (шейный отдел); с 22 лет — множественный кариес. С 26-летнего возраста стала ощущать боли в пояснице, которые постепенно нарастали, появились затруднения при ходьбе (скованность мышц нижних конечностей). Присоединилось онемение верхних и нижних конечностей после стресса, онемение тела усиливалось во время сна. Во время первой и второй беременности отмечала ухудшение: усилилась боль в сердце и в области поясницы. Дети шести и четырех лет до настоящего момента здоровы.
Осмотр пациентки показал следующее: состояние удовлетворительное, определяется сколиоз в шейном и поясничном отделах позвоночника. АД 140/90 мм рт.ст., тоны сердца приглушены, экстрасистолия, ЧСС 92 удара в 1 минуту, живот при пальпации мягкий, безболезненный.
Status localis: на коже туловища розовые шелушащиеся пятна, сухость кожных покровов, чешуйки снимаются с трудом. В области больших складок гиперкератоз, однако наиболее выражен в области нижних конечностей, ногтевые пластинки деформированы, изменен их цвет. Лицо не поражено. Пузыри появляются в жаркую погоду в области конечностей и шеи. Волосы, брови и ресницы не поражены.
Осмотр невролога: астенизирована, эмоционально лабильна, супраорбитальные точки болезненные, боль при движении глазными яблоками, склеры инъецированы, сглажена правая носогубная складка, положительный симптом Маринеску — Радовича с двух сторон. Сухожильные рефлексы оживлены с верхних и нижних конечностей, D ≥ S. Патологических стопных знаков нет. Положительный симптом Нери и Лассега под углом 70–80 градусов с двух сторон. Снижена болевая чувствительность в правой половине тела.
У пациентки имеют место следующие неврологические синдромы: церебрастенический, цефалгический, обусловленный венозно-ликворной дисциркуляцией, недостаточность функции левого лицевого нерва по центральному типу, правосторонняя пирамидная недостаточность, рефлексы орального автоматизма, рефлекторная цервикалгия, люмбалгия.
Локализация патологического процесса — базально-стволовые отделы головного мозга, спинномозговые нервы цервикального и люмбального уровней.
Характер церебрального процесса — сосудистый, в виде хронической цереброваскулярной недостаточности, развившейся на фоне краниовертебральной аномалии Арнольда — Киари 1, ретролистеза С3 II ст. с компрессией сосудов шеи, возможно, сопутствующего васкулита сосудов головного мозга, а также гипоксического характера в связи с сопутствующей малой аномалией сердца.
Заключение
Венозная энцефалопатия II на фоне аномалии Арнольда — Киари 1 и шейного остеохондроза с ретролистезом С3 II степени, умеренный стойкий болевой синдром. Вертеброгенная люмбоишиалгия. МРТ головного мозга: множественные гиперинтенсивные мелкие очаги, расположенные перивентрикулярно. Умеренная наружная гидроцефалия. МРТ шейного отдела позвоночника: аномалии Арнольда — Киари 1, шейный остеохондроз, спондилоартроз в сегментах С2-С3, С3-С4 , ретролистез тела С3 второй степени.
Консультация кардиолога: дисметаболическая кардиомиопатия. Гипертоническая болезнь II степени. Нарушение ритма (экстрасистолия), СН — 1, ФК — 2.
Малая аномалия сердца: атипичная хорда левого желудочка.
Практический интерес данных клинических случаев состоит в том, что врожденный ихтиоз выявлен у пациентов в нескольких поколениях. Он характеризуется сочетанием кожной симптоматики с патологией костной системы, опорно-двигательного аппарата, поражением центральной нервной системы, обусловленным системным васкулитом в периферических сосудах, сосудов головного и спинного мозга, почек, врожденными аномалиями сердца.
Таким образом, тяжесть состояния больных при ихтиозе зависит не только от поражения кожных покровов, но и от сопутствующей патологии, связанной с недоразвитием, пороками внутренних органов, нервной системы, что, по-видимому, обусловлено мутацией генов.