Статья опубликована на с. 159-162
Существует несколько определений болезни Фара, описанной T. Fahr в 1930 г. [3, 6, 17]:
1. Болезнь Фара — стриато-паллидо-дентатный кальциноз, феррокальциноз, цереброваскулярная интракраниальная кальцификация, минерализующая микроангиопатия.
2. Неатеросклеротическое обызвествление коры полушарий, базальных ганглиев и зубчатых ядер мозжечка, связанное с отложением солей кальция и железа в стенки мелких артерий и артериол, а также в вещество головного мозга [12].
3. Болезнь Фара — это нейродегенеративное заболевание, сопровождающееся идиопатической неатеросклеротической симметричной интрацеребральной кальцификацией базальных ганглиев [5].
Нередко в клинической картине болезни встречается первичное (чаще аутоиммунное) или послеоперационное нарушение функции паращитовидной железы. В этих случаях изменение выработки паратгормона приводит к снижению содержания кальция и повышению уровня фосфата в крови. Описаны случаи псевдогипопаратиреоза при легочной патологии, когда вследствие гипервентиляции и гипокапнии увеличивается внутриклеточное содержание фосфора [14]. При проведениии рентгенографии черепа или компьютерной томографии (КТ) у пациентов с болезнью Фара в головном мозге наблюдаются множественные симметричные очаги обызвествления (рентгенологический синдром Фара).
Гипоксией объясняют кальцификацию мозга при гипертонической болезни. Описан семейный случай заболевания с явлением генетической антиципации, при этом авторам впервые удалось идентифицировать хромосомный локус болезни Фара [7]. Дифференциальная диагностика болезни Фара чаще проводится с паразитарным поражением нервной системы (токсоплазмоз, эхинококкоз, цистицеркоз). В этих случаях диагностическую ценность представляют эпидемиологический анамнез и специфические серологические реакции в крови и спинномозговой жидкости. Реже заболевание дифференцируют с туберозным склерозом Бурневилля, при котором встречаются характерные изменения кожи (гипопигментация).
Семейные случаи первичной кальцификации базальных ганглиев, возникающие в отсутствие какого-либо другого заболевания и проявляющиеся паркинсонизмом, дистонией, деменцией, эпилептическими припадками, мозжечковыми и пирамидными симптомами, обозначаются как семейный стриато-паллидо-дентатный кальциноз, или болезнь Фара [8, 9].
Нередко отмечаются проявления гипер- или гипопаратиреоза: локальные судороги, тетанические спазмы, боли в дистальных отделах конечностей, положительные симптомы Хвостека и Труссо. Условно выделяют три группы больных: лиц молодого возраста с признаками церебрального кальциноза, пациентов с гипопаратиреозом и пожилых больных с относительно невыраженной кальцификацией [1, 2, 4].
Заболевание характеризуется симметричной кальцификацией вещества головного мозга, прежде всего в области базальных ганглиев и зубчатых ядер мозжечка. В тяжелых случаях кальцификации подвергаются таламус, кора больших полушарий и мозжечка, белое вещество в семиовальном центре [10–12].
Выделяют ювенильную и сенильную формы заболевания. Ювенильная форма проявляется у детей и подростков хореей или хореоатетозом, дистонией, дизартрией, эпилептическими припадками. У некоторых больных отмечается умственная отсталость, другие — интеллектуально сохранны. С возрастом гиперкинезы могут замещаться паркинсоническими симптомами. Основным проявлением сенильной формы, наблюдающейся у лиц среднего и пожилого возраста, считается паркинсонизм, который проявляется гипокинезией, ригидностью, микробазией, флексорной позой, замедленной монотонной речью, может сопровождаться другими экстрапирамидными синдромами, деменцией подкорково-лобного типа, мозжечковой атаксией, реже — пирамидной недостаточностью, эпилептическими припадками, недержанием мочи [13, 14].
При недостаточной деятельности паращитовидных желез при болезни Фара в результате гипокальциемии возможно развитие катаракты [6]. Помутнение хрусталика при тетании может возникнуть в течение нескольких часов. При биомикроскопии в корковом слое хрусталика, под передней и задней капсулами видны точечные и штрихообразные помутнения серого цвета, перемежающиеся с вакуолями и водными щелями, в дальнейшем катаракта прогрессирует [15, 16].
Гемиатрофия тела отмечается с детства и проявляется асимметрией кистей, стоп (различный размер обуви с двух сторон), лица, асимметричной гипотрофией мышц туловища и конечностей. В большинстве случаев костно-мышечная асимметрия выражена умеренно и сама по себе не приводит к серьезному ограничению движений [17].
Экстрапирамидные симптомы манифестируют, как правило, на 4–5-м десятилетии жизни. У больных постепенно развивается клиническая картина гемипаркинсонизма с тремором и мышечной ригидностью в конечностях на стороне гемиатрофии. Весьма типичным и ранним симптомом является дистония в руке и ноге, усиливающаяся при движении и не связанная с назначением препаратов леводопы. В некоторых случаях дистония может быть первичным неврологическим проявлением заболевания. Характерной особенностью данного синдрома является строгая односторонность симптоматики на протяжении многих лет и десятилетий от момента дебюта экстрапирамидных проявлений, что отличает синдром гемипаркинсонизма-гемиатрофии от болезни Паркинсона. На поздней стадии заболевания симптомы паркинсонизма могут отмечаться и на противоположной стороне, однако их выраженность всегда бывает намного ниже, чем на стороне исходной локализации гемипаркинсонизма [17].
При КТ и магнитно-резонансной томографии (МРТ) головного мозга у больных могут выявляться признаки утолщения костей свода черепа, расширение придаточных пазух носа, расширение бокового желудочка и корковых борозд на стороне, противоположной гемипаркинсонизму-гемиатрофии. Но в ряде случаев методы нейровизуализации не выявляют отклонений. При позитронно-эмиссионной томографии обнаруживается снижение метаболизма глюкозы в области базальных ганглиев и лобной коры. Указанные изменения могут быть двусторонними, но при этом они значительно более выражены в полушарии, контралатеральном гемипаркинсонизму.
Синдром гемипаркинсонизма-гемиатрофии отличается от болезни Паркинсона сравнительно благоприятным течением и медленным прогрессированием. Длительность заболевания может достигать 30 и более лет. В большинстве случаев назначение препаратов леводопы эффективно лишь в начальной стадии болезни, в то время как по мере прогрессирования заболевания их эффект заметно снижается.
Диагноз устанавливается с помощью КТ, выявляющей гиперденсивные изменения в области базальных ганглиев и зубчатых ядер мозжечка, иногда в других отделах мозга. МРТ значительно хуже выявляет кальцификацию, чем КТ. На Т2-взвешенных изображениях зона кальцификации выявляется в зависимости от концентрации кальция, химической структуры кальциевых отложений и наличия сопутствующих дегенеративных изменений.
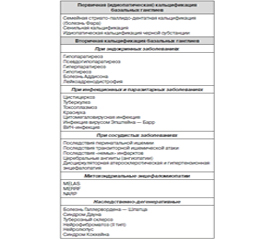
Причина кальцификации вещества мозга остается неясной, но все же основные этиологические факторы идентифицированы и представлены в табл. 1.
К сожалению, не всегда при лабораторном исследовании выявляются изменения показателей обмена кальция, фосфора и паратгормона. У внешне здоровых родственников больных КТ может выявлять асимптомную кальцификацию базальных ганглиев. Необходимо длительно проводить дифференциальную диагностику с другими заболеваниями, вызывающими внутримозговую кальцификацию, ведь лечение болезни Фара носит преимущественно симптоматический характер [18, 19].
Значительно чаще встречается вторичная кальцификация, которая может быть вызвана различными заболеваниями и иногда обозначается как синдром Фара. В большинстве случаев она связана с гипопаратиреозом или псевдогипопаратиреозом.
Гипопаратиреоз возникает вследствие уменьшения секреции гормона паращитовидных желез (паратгормона), что вызывает снижение содержания в крови кальция и повышение уровня фосфатов. Дефицит гормона обычно бывает результатом тиреоидэктомии, реже — идиопатического заболевания паращитовидной железы.
Псевдогипопаратиреоз — наследственное заболевание, передающееся по доминантному сцепленному с полом типу (болеют преимущественно мальчики). Клинически оно сходно с гипопаратиреозом и тоже сопровождается снижением содержания кальция и повышением содержания фосфора в крови, но возникает из-за нечувствительности периферических тканей к действию паратгормона. У больных часто отмечаются округлое лицо, укорочение костей запястья и предплюсны, умственная отсталость.
При проведении патологоанатомического исследования макроскопически в головном мозге часто наблюдаются участки с ветвистыми плотными белесоватыми сосудами, издающими под лезвием ножа дробный хруст. При гистологическом исследовании срезов головного мозга (чаще — коры полушарий, мозжечка, базальных ганглиев) характерным является обнаружение кольцевидных отложений солей кальция, расположенных между адвентицией сосудов и средней оболочкой или пограничной мембраной глии.
Кальцификация базальных ганглиев выявляется у 70 % больных с идиопатическим гипопаратиреозом и почти у всех больных с псевдогипопаратиреозом. Чаще всего кальцификация бывает асимптомной и лишь у небольшой части больных сопровождается развитием паркинсонизма, обычно на фоне более распространенного неврологического дефекта. У части больных развиваются дистония, хореоатетоз или тяжелый постурально-кинетический тремор.
И все же клиницисту (неврологу, эндокринологу, семейному врачу, окулисту) в первую очередь обязательно обследовать больных на наличие причин нарушения обмена кальция.
Как при гипопаратиреозе, так и при псевдогипопаратиреозе экстрапирамидные синдромы часто сопровождаются тетанией, парестезиями в дистальных отделах конечностей и губ, судорожными припадками, умственной отсталостью или деменцией. Судороги редко имеют симптоматический характер и обычно не реагируют на антиконвульсанты, но регрессируют после нормализации уровня кальция. У части больных развивается внутричерепная гипертензия с отеком дисков зрительных нервов, которая тоже регрессирует после коррекции уровня кальция в крови. При осмотре выявляются признаки повышенной нервно-мышечной возбудимости (тетании) — симптомы Хвостека и Труссо, карпопедальные спазмы, снижение сухожильных рефлексов.
У части больных симптомы паркинсонизма, как и другие клинические проявления, улучшаются на фоне коррекции гипокальциемии (при комбинированной терапии препаратами витамина D (кальций D3)). Препараты леводопы обычно малоэффективны.
Сходную клиническую картину может вызывать псевдогипопаратиреоз — заболевание неясной этиологии, клинически напоминающее гипопаратиреоз и псевдогипопаратиреоз (тетания, судорожные припадки, снижение интеллекта, деформация скелета, кальцификация базальных ганглиев и т.д.), но не сопровождающееся гипокальциемией или гиперфосфатемией.
Описаны случаи избирательной кальцификации черной субстанции неясного генеза, сопровождающиеся паркинсонизмом. Возможно сочетание стриато-паллидо-дентатного кальциноза с боковым амиотрофическим склерозом, прогрессирующим надъядерным параличом.
И все же больные с начальными проявлениями нарушения обмена кальция обращаются к неврологу. В связи с этим при первичном обследовании крайне необходимо исследование обмена кальция, а возможно, и последующее динамическое повторение обследования. Болезнь Фара — редкое (орфанное) заболевание, и выявляют ее достаточно длительное течение на КТ — четко дифференцируют наличие симметричных кальцинатов.
Лечение продолжает быть симптоматичным [2]. Дистонические атаки — прегабалин (габапентин), тенотен, магний В6 + кальций D3 или гамалате В6 + кальций D3, при нарушении сна — мелатонин. Целесообразно применение курсами левокарнитина (агвантара), стимол + коэнзим. В связи со снижением когниций подобная терапия проводится 4 раза в год, в промежутке — семакс (дельталицин) капли в нос + гинко билоба, цитофлавин внутривенно с последующей таблетированной формой. Лечение при хорошей переносимости расписывается на целый год.
Список литературы
1. Величко М.А., Васильев В.В., Филиппов Ю.Л. Синдром Фара при гипертонической болезни // Клиническая медицина. — 1993. — № 2. — С. 55-58.
2. Евтушенко С.К. Синдром болезни Фара (соматоневрологические проявления) // Редкие и труднодиагностируемые заболевания нервной системы. — Святогорск, 2003. — С. 15-20.
3. Петелин Л.С., Фокин М.А., Борзуновса Т.А., Шаповалова М.В. Синдром Фара // Журнал неврологии и психиатрии. — 1988. — № 9. — С. 65-67.
4. Пономарев В.В., Науменко Д.В. Болезнь Фара: клиническая картина и подходы к лечению // Журнал неврологии и психиатрии. — 2004. — № 3. — С. 42-45.
5. Федулова М.В., Русакова Т.И., Ермоленко Э.Н. Болезнь Фара, выявленная при судебно-медиицнской экспертизе // Судебно-медицинская экспертиза. — 2006. — № 5. — С. 35-37.
6. Трухан Д.И., Лебедов О.И. Изменения органа зрения при заболеваниях внутренних органов. — М., 2014. — 208 с.
7. Яхно Н.Н. Болезнь Фара // Вопросник неврологический. — 2001. — № 4. — С. 17-22.
8. Fahr T. // Zbl. Allg. Pathol. — 1930. — Bd. 30. — S. 129-133.
9. Gescywind D.H., Loginov M., Stern J.M. et al. Identification of a locus on chromosome 14q for idiopathic basal ganglia calcification // Am. J. Hum. Genet. — 2009. — S. 764-772.
10. Guseo A., Boldizsar R., Gellert et al. Electron microscopic study of striatodental calcification // Acta neuropathology. — 1975. — № 4. — S. 305-331.
11. Gescnwind D.H., Loginov M. Identification of a locus on chromosome 14q for idiopathic basal ganglia calcification // Am. J. Hum. Genet. — 2005. — № 65(3). — Р. 764-772.
12. Goldscheider H.G., Lischewski R. Clinical, endocrinologicakl, and computerized tomography scans for symmetrical calcification of the basal ganglia // Arch. Psychiat. Nervenktn. — 1980. — № 228(1). — Р. 53-65.
13. Cyseo A., Boldizsar F. Electron microscopic study of striatodental calcification // Acta Neuropathol. — 2006. — № 31(4). — Р. 305-313.
14. Maghraoul A., Birouk N. Fahr syndrome and dysparathytoidism // Presse Med. — 1995. — № 24(28). — 1301-1304.
15. Proniska E., Kulczycki J., Rovinska E., Kuran W. Abolished phosphaturic response to parathormone in adult patients with Fahr disease and its restoration after propranolol administration // J. Neurol. — 1988. — № 235(3). — Р. 185-187.
16. Rossi N., Morena M. Calcification of the basal ganglia and Fahr disease. Report of two clinical cases and review of the literature // Recenti Prog. Med. — 2009. — № 84(3). — Р. 192-198.
17. Stellamor K., Stellamor V. Roentgen diagnosis og Fahr’s disease // Rontgenblatter. — 1983. — № 36(6). — Р. 194-11.
18. Tardio E., Roldan M.L., Pedrola D., Hierro F.R. Fahr disease and idiopathic pulmonary hemosiderosis in a 10 year old patient // An. Esp. Pediat. — 1980. — № 13(7). — Р. 599-604.
19. Taxer F., Haller R. Clinical early symptoms and CT findigs in Fahr syndrome // Nervenarzt. — 2006. — № 57(10). — Р. 583-588.

/161.jpg)