
Международный эндокринологический журнал Том 18, №3, 2022
Вернуться к номеру
Епігенетика, клітинний цикл та метаболізм стовбурових клітин. Формування інсулін-продукуючих клітин
Авторы: Тронько М.Д., Пушкарьов В.М., Ковзун О.І., Соколова Л.К., Пушкарьов В.В.
ДУ «Інститут ендокринології та обміну речовин ім. В.П. Комісаренка НАМН України», м. Київ, Україна
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
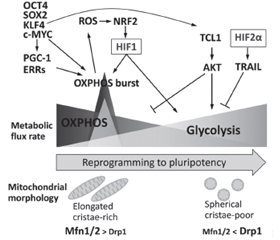
Диференціація стовбурових клітин (SC) вимагає низки перебудов хроматину для встановлення клітинної ідентичності. Посттрансляційні модифікації гістонів зазвичай регулюють динаміку гетерохроматину. Гістони піддаються різним модифікаціям, таким як ацетилювання, метилювання, фосфорилювання та убіквітинування, і таким чином сприяють регуляції стану хроматину та транскрипційній активності. Хімічно стабільний патерн метильованих гістонів сприяє клітинній пам’яті щодо зовнішніх стимулів, підтримуючи рівні транскрипції адаптивних генів навіть після усунення сигналів навколишнього середовища. Модифікації хроматину відіграють важливу роль у дозріванні клітин острівців підшлункової залози, встановленні схеми секреції, яка стимулює регуляцію секреції інсуліну. МікроРНК, клас ендогенних малих некодуючих РНК в еукаріотів, є важливими регуляторами експресії генів на рівні посттранскрипційних механізмів. МікроРНК регулюють секрецію інсуліну, розвиток підшлункової залози та диференціювання β-клітин. Плюрипотентні SC характеризуються високою швидкістю проліферації, здатністю до самовідновлення та потенціалом диференціації у різні типи клітин. Ця швидка проліферація зумовлена модифікованим клітинним циклом, який дозволяє клітинам швидко переходити від синтезу ДНК до поділу клітини за рахунок скорочення часу проміжних (G1 і G2) фаз. Канонічний сигнальний шлях WNT/β-катеніну характеризується як основний драйвер росту та проліферації клітин. Під час G1 сигналінг WNT індукує перехід до S-фази. Порівняно з їхніми соматичними аналогами плюрипотентні SC демонструють високу швидкість гліколізу, подібну до аеробного гліколізу в ракових клітинах, — явище, відоме як ефект Варбурга, яке є важливим для підтримки властивостей SC. У стовбурових клітинах позаклітинне надходження Ca2+ в цитоплазму опосередковується головним чином депо-керованими Ca2+-каналами. Показано, що позаклітинний кальцій сприяє проліферації SC, а отже, може брати участь в трансплантаційній терапії.
Stem cell (SC) differentiation requires a series of chromatin rearrangements to establish cell identity. Posttranslational modifications of histones usually regulate the dynamics of heterochromatin. Histones are subjected to various modifications, such as acetylation, methylation, phosphorylation and ubiquinination, and thus contribute to regulation of chromatin status and transcriptional activity. The chemically stable pattern of methylated histones promotes cellular memory relative to external stimuli, maintaining transcription levels of adaptive genes even after elimination of environmental signals. Chromatin modifications play an important role in the maturation of pancreatic islet cells, the establishment of a secretion pattern that stimulates the regulation of insulin secretion. MicroRNAs, a class of endogenous small noncoding RNAs in eukaryotes, are important regulators of gene expression at the level of posttranscriptional mechanisms. MicroRNAs regulate insulin secretion, pancreatic development, and β-cell differentiation. Pluripotent SCs are characterized by a high rate of proliferation, the ability to self-repair and the potential for differentiation in different cell types. This rapid proliferation is due to a modified cell cycle that allows cells to rapidly transition from DNA synthesis to cell division by reducing the time of gap (G1 and G2) phases. The canonical WNT/β-catenin signaling pathway is characterized as a major driver of cell growth and proliferation. At G1, WNT signaling induces a transition to the S-phase. Compared to their somatic counterparts, pluripotent SCs exhibit a high rate of glycolysis similar to aerobic glycolysis in cancer cells, a phenomenon known as the Warburg effect, which is important for maintaining SC properties. In stem cells, the extracellular influx of Ca2+ into the cytoplasm is mediated mainly by depot-controlled Ca2+ channels. Extracellular calcium has been shown to promote SC proliferation and thus may be involved in transplant therapy.
стовбурові клітини; епігенетичні модифікації; клітинний цикл; метаболізм; іони кальцію; інсулін-продукуючі клітини
stem cells; epigenetic modifications; cell cycle; metabolism; calcium ions; insulin-producing cells
Епігенетика стовбурових клітин
Особливості клітинного циклу стовбурових клітин
/39.jpg)
Клітинний цикл і диференціація плюрипотентних стовбурових клітин
Потенційна роль WNT у регулюванні фази G1 PSC
/41.jpg)
Роль іонів кальцію у функціонуванні стовбурових клітин
Участь Ca2+ у диференціації SC
Адгезія, міграція та хомінг
Старіння SC
Висновки
- Krentz N.A.J., Gloyn A.L. Insights into pancreatic islet cell dysfunction from type 2 diabetes mellitus genetics. Nat. Rev. Endocrinol. 2020. 16. 202-12. doi: 10.1038/s41574-020-0325-0.
- Kampmann M. CRISPR-based functional genomics for neurological disease. Nat. Rev. Neurol. 2020. 16(9). 465-80. doi: 10.1038/s41582-020-0373-z.
- Astro V., Adamo A. Epigenetic Control of Endocrine Pancreas Differentiation in vitro: Current Knowledge and Future Perspectives. Front Cell Dev. Biol. 2018. 6. 141. doi: 10.3389/fcell.2018.00141.
- Nicetto D., Donahue G., Jain T., Peng T., Sidoli S., Sheng L., et al. H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification. Science. 2019. 363(6424). 294-7. doi: 10.1126/science.aau0583.
- Thienpont B., Aronsen J.M., Robinson E.L., Okkenhaug H., Loche E., Ferrini A., et al. The H3K9 dimethyltransferases EHMT1/2 protect against pathological cardiac hypertrophy. J. Clin. Invest. 2017. 127(1). 335-48. doi: 10.1172/JCI88353.
- Strom A.R., Emelyanov A.V., Mir M., Fyodorov D.V., Darzacq X., Karpen G.H. Phase separation drives heterochromatin domain formation. Nature. 2017. 547. 241-5. doi: 10.1038/nature22989.
- Ninova M., Fejes Tóth K., Aravin A.A. The control of gene expression and cell identity by H3K9 trimethylation. Development. 2019. 146(19). dev181180. doi: 10.1242/dev.181180.
- Lu T.T., Heyne S., Dror E., Casas E., Leonhardt L., Boenke T., et al. The Polycomb-Dependent Epigenome Controls β Cell Dysfunction, Dedifferentiation, and Diabetes. Cell Metab. 2018. 27(6). 1294-308.e7. doi: 10.1016/j.cmet.2018.04.013.
- Lee D.H., Kim G.W., Jeon Y.H., Yoo J., Lee S.W., Kwon S.H. Advances in histone demethylase KDM4 as cancer therapeutic targets. FASEB J. 2020. 34(3). 3461-84. doi: 10.1096/fj.201902584R.
- Rosales W., Lizcano F. The Histone Demethylase JMJD2A Modulates the Induction of Hypertrophy Markers in iPSC-Derived Cardiomyocytes. Front Genet. 2018. 9. 14. doi: 10.3389/fgene.2018.00014.
- Arroyave F., Montaño D., Lizcano F. Diabetes Mellitus Is a Chronic Disease that Can Benefit from Therapy with Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2020. 21(22). 8685. doi: 10.3390/ijms21228685.
- Zhang T., Huang K., Zhu Y., Wang T., Shan Y., Long B., et al. Vitamin C-dependent lysine demethylase 6 (KDM6)-mediated demethylation promotes a chromatin state that supports the endothelial-to-hematopoietic transition. J. Biol. Chem. 2019. 294(37). 13657-70. doi: 10.1074/jbc.RA119.009757.
- Coskun E., Ercin M., Gezginci-Oktayoglu S. The Role of Epigenetic Regulation and Pluripotency-Related MicroRNAs in Differentiation of Pancreatic Stem Cells to Beta Cells. J. Cell Biochem. 2018. 119(1). 455-67. doi: 10.1002/jcb.26203.
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006. 126(4). 663-76. doi: 10.1016/j.cell.2006.07.024.
- Liu X., Ouyang J.F., Rossello F.J., Tan J.P., Davidson K.C., Valdes D.S., et al. Reprogramming roadmap reveals route to human induced trophoblast stem cells. Nature. 2020. 586. 101-107. doi: 10.1038/s41586-020-2734-6.
- Wang Y., Bi Y., Gao S. Epigenetic regulation of somatic cell reprogramming. Curr. Opin. Genet. Dev. 2017. 46. 156-163. doi: 10.1016/j.gde.2017. 07.002.
- Di Stefano M., Stadhouders R., Farabella I., Castillo D., Serra F., Graf T., et al. Transcriptional activation during cell reprogramming correlates with the formation of 3D open chromatin hubs. Nat. Commun. 2020. 11. 2564. doi: 10.1038/s41467-020-16396-1.
- Lu L., Liu X., Huang W.K., Giusti-Rodriguez P., Cui J., Zhang S., et al. Robust Hi-C Maps of Enhancer-Promoter Interactions Reveal the Function of Non-coding Genome in Neural Development and Diseases. Mol. Cell. 2020. 79. 521-534e515. doi: 10.1016/j.molcel.2020.06.007.
- Stadhouders R., Vidal E., Serra F., Di Stefano B., Le Dily F., Quilez J., et al. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat. Genet. 2018. 50. 238-249. doi: 10.1038/s41588-017-0030-7.
- Li D., Liu J., Yang X., Zhou C., Guo J., Wu C., et al. Chromatin Accessibility Dynamics during iPSC Reprogramming. Cell Stem Cell. 2017. 21. 819-833e816. doi: 10.1016/j.stem.2017.10.012.
- Sun L., Fu X., Ma G., Hutchins A.P. Chromatin and Epigenetic Rearrangements in Embryonic Stem Cell Fate Transitions. Front. Cell Dev. Biol. 2021. 9. 637309. doi: 10.3389/fcell.2021.637309.
- Chen J., Guo L., Zhang L., Wu H., Yang J., Liu H., et al. Vitamin C modulates TET1 function during somatic cell reprogramming. Nat. Genet. 2013. 45. 1504-1509. doi: 10.1038/ng.2807.
- Arabaci D.H., Terzioglu G., Bayirbasi B., Onder T.T. Going up the Hill: Chromatin-based Barriers to Epigenetic Reprogramming. FEBS J. 2020. 15628. doi: 10.1111/febs.15628.
- Chen J., Liu H., Liu J., Qi J., Wei B., Yang J., et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 2013. 45. 34-42. doi: 10.1038/ng.2491.
- Miles D.C., de Vries N.A., Gisler S., Lieftink C., Akhtar W., Gogola E., et al. TRIM28 is an Epigenetic Barrier to Induced Pluripotent Stem Cell Reprogramming. Stem Cells. 2017. 35. 147-157. doi: 10.1002/stem.2453.
- Klimczak M., Czerwinska P., Mazurek S., Sozanska B., Biecek P., Mackiewicz A., et al. TRIM28 epigenetic corepressor is indispensable for stable induced pluripotent stem cell formation. Stem Cell Res. 2017. 23. 163-172. doi: 10.1016/j.scr. 2017.07.012.
- Chronis C., Fiziev P., Papp B., Butz S., Bonora G., Sabri S., et al. Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell. 2017. 168. 442-459e420. doi: 10.1016/j.cell.2016.12.016.
- Zhuang Q., Li W., Benda C., Huang Z., Ahmed T., Liu P., et al. NCoR/SMRT co-repressors cooperate with c-MYC to create an epigenetic barrier to somatic cell reprogramming. Nat. Cell Biol. 2018. 20. 400-12. doi: 10.1038/ s41556-018-0047-x.
- Huang Y., Zhang H., Wang L., Tang C., Qin X., Wu X., et al. JMJD3 acts in tandem with KLF4 to facilitate reprogramming to pluripotency. Nat. Commun. 2020. 11. 5061. doi: 10.1038/s41467-020-18900-z.
- Rao R.A., Dhele N., Cheemadan S., Ketkar A., Jayandharan G.R., Palakodeti D., et al. Ezh2 mediated H3K27me3 activity facilitates somatic transition during human pluripotent reprogramming. Sci. Rep. 2015. 5. 8229. doi: 10.1038/ srep08229.
- Li H., Lai P., Jia J., Song Y., Xia Q., Huang K., et al. RNA Helicase DDX5 Inhibits Reprogramming to Pluripotency by miRNA-Based Repression of RYBP and its PRC1-Dependent and Independent Functions. Cell Stem Cell. 2017. 20. 571. doi: 10.1016/j.stem.2017.03.014.
- Sun Z.Y., Yu T.Y., Jiang F.X., Wang W. Functional maturation of immature β cells: A roadblock for stem cell therapy for type 1 diabetes. World J. Stem Cells. 2021. 13(3). 193-207. doi: 10.4252/wjsc.v13.i3.193.
- Boward B., Wu T., Dalton S. Concise Review: Control of Cell Fate through Cell Cycle and Pluripotency Networks. Stem Cells. 2016. 34(6). 1427-36. doi: 10.1002/stem.2345.
- Rasmussen M.L., Ortolano N.A., Romero-Morales A.I., Gama V. Wnt Signaling and It’s Impact on Mitochondrial and Cell Cycle Dynamics in Pluripotent Stem Cells. Genes (Basel). 2018. 9(2). 109. doi: 10.3390/genes9020109.
- Zaveri L., Dhawan J. Cycling to Meet Fate: Connecting Pluripotency to the Cell Cycle. Front Cell Dev. Biol. 2018. 6. 57. doi: 10.3389/fcell.2018.00057.
- Kolupaeva V., Janssens V. PP1 and PP2A phosphatases-cooperating partners in modulating retinoblastoma protein activation. FEBS J. 2013. 280(2). 627-43. doi: 10.1111/j.1742-4658.2012.08511.x.
- Bertoli C., Skotheim J.M., de Bruin R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013. 14(8). 518-28. doi: 10.1038/nrm3629.
- Soufi A., Dalton S. Cycling through developmental decisions: how cell cycle dynamics control pluripotency, differentiation and reprogramming. Development. 2016. 143(23). 4301-11. doi: 10.1242/dev.142075.
- Ter Huurne M., Chappell J., Dalton S., Stunnenberg H.G. Distinct Cell-Cycle Control in Two Different States of Mouse Pluripotency. Cell Stem Cell. 2017. 21(4). 449-55.e4. doi: 10.1016/j.stem.2017.09.004.
- Pauklin S., Vallier L. The cell-cycle state of stem cells determines cell fate propensity. Cell. 2013. 155(1). 135-47. doi: 10.1016/j.cell.2013.08.031. Erratum in: Cell. 2014. 156(6). 1338.
- Pauklin S., Madrigal P., Bertero A., Vallier L. Initiation of stem cell differentiation involves cell cycle-dependent regulation of developmental genes by Cyclin D. Genes. Dev. 2016. 30(4). 421-33. doi: 10.1101/gad.271452.115.
- Liu L., Michowski W., Inuzuka H., Shimizu K., Nihira N.T., Chick J.M., et al. G1 cyclins link proliferation, pluripotency and differentiation of embryonic stem cells. Nat. Cell Biol. 2017. 19(3). 177-88. doi: 10.1038/ncb3474.
- Roccio M., Schmitter D., Knobloch M., Okawa Y., Sage D., Lutolf M.P. Predicting stem cell fate changes by differential cell cycle progression patterns. Development. 2013. 140(2). 459-70. doi: 10.1242/dev.086215.
- Ponti G., Obernier K., Guinto C., Jose L., Bonfanti L., Alvarez-Buylla A. Cell cycle and lineage progression of neural progenitors in the ventricular-subventricular zones of adult mice. Proc. Natl. Acad. Sci. USA. 2013. 110(11). E1045-54. doi: 10.1073/pnas.1219563110.
- Pauklin S., Vallier L. The cell-cycle state of stem cells determines cell fate propensity. Cell. 2013. 155(1). 135-47. doi: 10.1016/j.cell.2013.08.031.
- Davidson G. The cell cycle and Wnt. Cell Cycle. 2014. 9. 1667-8. doi: 10.4161/cc.9.9.11595.
- De Jaime-Soguero A., Aulicino F., Ertaylan G., Griego A., Cerrato A., Tallam A., et al. Wnt/Tcf1 pathway restricts embryonic stem cell cycle through activation of the Ink4/Arf locus. PLoS Genet. 2017. 13(3). e1006682. doi: 10.1371/journal.pgen.1006682.
- Kim K.P., Han D.W., Kim J., Schöler H.R. Biological importance of OCT transcription factors in reprogramming and development. Exp. Mol. Med. 2021. 53(6). 1018-28. doi: 10.1038/s12276-021-00637-4.
- Ebrahimi A., Sevinç K., Gürhan Sevinç G., Cribbs A., Philpott M., Uyulur F., et al. Bromodomain inhibition of the coactivators CBP/EP300 facilitate cellular reprogramming. Nat. Chem. Biol. 2019. 15. 519-28. doi: 10.1038/s41589-019-0264-z.
- Kim K.P., Choi J., Yoon J., Bruder J.M., Shin B., Kim J., et al. Permissive epigenomes endow reprogramming competence to transcriptional regulators. Nat. Chem. Biol. 2021. 17. 47-56. doi: 10.1038/s41589-020-0618-6.
- Kim K.P., Wu Y., Yoon J., Adachi K., Wu G., Velychko S., et al. Reprogramming competence of OCT factors is determined by transactivation domains. Sci. Adv. 2020. 6(36). eaaz7364. doi: 10.1126/sciadv.aaz7364.
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Sasaki A., Yamamoto M., et al. Induction of pluripotency in human somatic cells via a transient state resembling primitive streak-like mesendoderm. Nat. Commun. 2014. 5. 3678. doi: 10.1038/ncomms4678.
- Ishida T., Nakao S., Ueyama T., Harada Y., Kawamura T. Metabolic remodeling during somatic cell reprogramming to induced pluripotent stem cells: involvement of hypoxia-inducible factor 1. Inflamm. Regen. 2020 May 12. 40. 8. doi: 10.1186/s41232-020-00117-8.
- Ahamad N., Singh B.B. Calcium channels and their role in regenerative medicine. World J. Stem Cells. 2021. 13(4). 260-80. doi: 10.4252/wjsc.v13.i4.260.
- Uzieliene I., Bernotas P., Mobasheri A., Bernotiene E. The Role of Physical Stimuli on Calcium Channels in Chondrogenic Differentiation of Mesenchymal Stem Cells. International Journal of Molecular Sciences. 2018. 19(10). 2998. https://doi.org/10.3390/ijms19102998.
- Uzieliene I., Bernotas P., Mobasheri A., Bernotiene E. The Role of Physical Stimuli on Calcium Channels in Chondrogenic Differentiation of Mesenchymal Stem Cells. Int. J. Mol. Sci. 2018. 19(10). 2998. doi: 10.3390/ijms19102998.
- Hao B., Webb S.E., Miller A.L., Yue J. The role of Ca(2+) signaling on the self-renewal and neural differentiation of embryonic stem cells (ESCs). Cell Calcium. 2016. 59(2-3). 67-74. doi: 10.1016/j.ceca.2016.01.004.
- Davenport B., Li Y., Heizer J.W., Schmitz C., Perraud A.L. Signature Channels of Excitability no More: L-Type Channels in Immune Cells. Front Immunol. 2015. 6. 375. doi: 10.3389/fimmu.2015.00375.
- Tan Y.Z., Fei D.D., He X.N., Dai J.M., Xu R.C., Xu X.Y., Wu J.J., Li B. L-type voltage-gated calcium channels in stem cells and tissue engineering. Cell Prolif. 2019. 52(4). e12623. doi: 10.1111/cpr.12623.
- Uslu M., Albayrak E., Kocabaş F. Temporal modulation of calcium sensing in hematopoietic stem cells is crucial for proper stem cell expansion and engraftment. J. Cell Physiol. 2020. 235(12). 9644-9666. doi: 10.1002/jcp.29777.
- Lee M.N., Hwang H.S., Oh S.H., Roshanzadeh A., Kim J.W., Song J.H., Kim E.S., Koh J.T. Elevated extracellular calcium ions promote proliferation and migration of mesenchymal stem cells via increasing osteopontin expression. Exp. Mol. Med. 2018. 50(11). 1-16. doi: 10.1038/s12276-018-0170-6.
- Liu M.Y., Hua W.K., Chiou Y.Y., Chen C.J., Yao C.L., Lai Y.T., Lin C.H., Lin W.J. Calcium-dependent methylation by PRMT1 promotes erythroid differentiation through the p38α MAPK pathway. FEBS Lett. 2020. 594(2). 301-316. doi: 10.1002/1873-3468.13614.
- Pchelintseva E., Djamgoz M.B.A. Mesenchymal stem cell differentiation: Control by calcium-activated potassium channels. J. Cell Physiol. 2018. 233(5). 3755-3768. doi: 10.1002/jcp.26120.
- Jiang L.H., Mousawi F., Yang X., Roger S. ATP-induced Ca2+-signalling mechanisms in the regulation of mesenchymal stem cell migration. Cell Mol. Life Sci. 2017. 74(20). 3697-3710. doi: 10.1007/s00018-017-2545-6.
- Ahamad N., Sun Y., Nascimento D., Conceicao V., Xavier Paul Ezhilan C.R.D., Natarajan M., Singh B.B. Differential activation of Ca2+ influx channels modulate stem cell potency, their proliferation/viability and tissue regeneration. NPJ Regen. Med. 2021. 6(1). 67. doi: 10.1038/s41536-021-00180-w.
- Ahamad N., Sun Y., Singh B.B. Increasing cytosolic Ca2+ levels restore cell proliferation and stem cell potency in aged MSCs. Stem Cell Res. 2021. 56. 102560. doi: 10.1016/j.scr.2021.102560.
- Jirawatnotai S., Dalton S., Wattanapanitch M. Role of cyclins and cyclin-dependent kinases in pluripotent stem cells and their potential as a therapeutic target. Semin Cell Dev. Biol. 2020. 107. 63-71. doi: 10.1016/j.semcdb.2020.05.001.