
Международный эндокринологический журнал Том 18, №3, 2022
Вернуться к номеру
Синдром Ларона: клініка, діагностика (клінічний випадок)
Авторы: Ляшук П.М. (1), Ляшук Р.П. (1), Станкова Н.І. (2), Кудіна М.Б. (2)
(1) — Буковинський державний медичний університет, м. Чернівці, Україна
(2) — Чернівецький обласний ендокринологічний центр, м. Чернівці, Україна
Рубрики: Эндокринология
Разделы: Справочник специалиста
Версия для печати
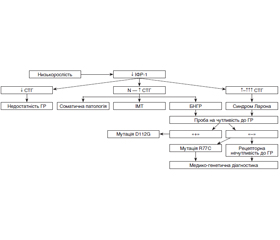
Поєднання нормального/високого рівня соматотропного гормону (СТГ) з низьким рівнем інсуліноподібного фактора росту-1 (ІФР-1) характерно для порушення рецепторної чутливості до СТГ — рідкісного генетично детермінованого синдрому, описаного ізраїльським клініцистом Z. Laron. Крім відносного дефіциту СТГ, на відміну від гіпофізарного нанізму, інші функції гіпофіза не змінені. На тлі різкого відставання в рості з раннього віку, іноді з внутрішньоутробного періоду, у дітей зберігаються нормальні пропорції тіла. Описано випадок рідкісної ендокринопатії — генетично детермінований синдром Ларона. Розглянуті етіологія, патогенез, особливості клінічного перебігу захворювання та перспективи лікувального підходу. Подано клінічні ознаки основних форм затримки росту та клінічний випадок. Діагноз синдрому Ларона встановлений на підставі низькорослості за відсутності проявів інших ендокринопатій, низького рівня ІФР-1 при нормальному референтному значенні СТГ крові та відсутності ефекту від лікування препаратом гормону росту. Пацієнтка потребує подальшого спостереження ендокринолога до періоду старту пубертату і за необхідності (у випадку формування комплексу неповноцінності) — психологічної корекції. Надані загальні рекомендації щодо повноцінності раціону харчування, режиму сну, фізичної активності та лікувальної фізкультури. Призначено полівітаміни. Обнадійливими є спроби застосування генно-інженерних препаратів ІФР-1. Сімейному лікарю та практикуючому ендокринологу при проведенні диференційної діагностики між основними формами затримки росту і низькорослості слід призначити гормональні дослідження крові та візуалізаційні методи обстеження відповідно до попереднього орієнтовного діагнозу за клінічними даними.
The combination of normal/high levels of somatotropic hormone with low levels of insulin-like growth factor-1 is characteristic of impaired receptor sensitivity to somatotropic hormone, a rare genetically determined syndrome described by Israeli clinician Z. Laron. In addition to the relative deficiency of somatotropic hormone, in contrast to pituitary dwarfism, other functions of the pituitary gland are not changed. Against the background of a sharp lag in growth from an early age, sometimes from the fetal period, children retain normal body proportions. The case of rare endocrinopathy — genetically determined Laron’s syndrome is described. Ethiopathogenesis, features of clinical course of disease and prospects of therapeutic approach are considered. The clinical signs of the main forms of growth retardation are presented. The diagnosis of Laron’s syndrome is made on the basis of short stature in the absence of other endocrinopathies, low levels of insulin-like growth factor-1 with a normal reference value of somatotropic hormone and no effect of treatment with somatotropic hormone. The patient needs further observation by an endocrinologist before the onset of puberty and, if necessary (in the case of the formation of inferiority complex), іn psychological correction. General recommendations on the completeness of the diet, sleep, physical activity and physical therapy are given. Prescribed multivitamins. Attempts to use insulin-like growth factor-1 genetically engineered drugs are encouraging. When making a differential diagnosis between the main forms of growth retardation and stunted growth, the family physician and endocrinologist should be prescribed hormonal blood tests and imaging methods according to the previous indicative diagnosis according to clinical data.
синдром Ларона; низькорослість; гормон росту; інсуліноподібний фактор росту; клініка; діагноз
Laron’s syndrome; low growth; growth hormone; insulin-like growth factor; clinic; diagnosis
Вступ
Опис клінічного випадку
/61.jpg)
Висновки
- Kim J.H., Chae H.W., Chin S.O., Ku C.R., Park K.H., Lim D.J., Kim K.J., et al. Diagnosis and Treatment of Growth Hormone Deficiency: A Position Statement from Korean Endocrine Society and Korean Society of Pediatric Endocrinology. Endocrinol. Metab. (Seoul). 2020. 35(2). 272-287. doi: 10.3803/EnM.2020.35.2.272.
- Laron Z. Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958-2003. J. Clin. Endocrinol. Metab. 2004. 89(3). 1031-44. doi: 10.1210/jc.2003-031033. PMID: 15001582.
- Kowarski A.A., Schneider J., Ben-Galim E., Weldon V.V., Daughaday W.H. Growth failure with normal serum RIA-GH and low somatomedin activity: somatomedin restoration and growth acceleration after exogenous GH. J. Clin. Endocrinol. Metab. 1978. 47(2). 461-4. doi: 10.1210/jcem-47-2-461. PMID: 263308.
- Walenkamp M.J., Wit J.M. Genetic disorders in the growth hormone — Insulin-like growth factor-I axis. Horm. Res. 2006. 66(5). 221-30. doi: 10.1159/000095161. PMID: 16917171.
- Takahashi Y., Kaji H., Okimura Y., Goji K., Abe H., Chihara K. Brief report: short stature caused by a mutant growth hormone. N. Engl. J. Med. 1996. 334(7). 432-6. doi: 10.1056/NEJM199602153340704.
- Kannenberg K., Wittekindt N.E., Tippmann S., Wolburg H., Ranke M.B., Binder G. Mutant and misfolded human growth hormone is rapidly degraded through the proteasomal degradation pathway in a cellular model for isolated growth hormone deficiency type II. J. Neuroendocrinol. 2007. 19(11). 882-90. doi: 10.1111/j.1365-2826.2007.01602.x. PMID: 17927666.
- Hage C., Gan H.W., Ibba A., Patti G., Dattani M., Loche S., Maghnie M., Salvatori R. Advances in differential diagnosis and management of growth hormone deficiency in children. Nat. Rev. Endocrinol. 2021. 17(10). 608-624. doi: 10.1038/s41574-021-00539-5.
- Liashuk P.M., Stankova N.I., Glugovska S.V., Lenkovska G.S. Congenital violations of sexual differentiation. International Journal of Endocrinology (Ukraine). 2013. 6(54). 112. (in Ukrainian)
- Liashuk P.M., Stankova N.I., Pashkovska N.V., Kryvych L.S. Nunen syndrome: a case study. Clinical and Experiment Pathology. 2011. 3(57). 156-7. (in Ukrainian)
- Liashuk P.M., Stankova N.O., Ilyushina A.A., Lyashuk R.P. Male Nunen syndrome. Clinical and Experiment Pathology. 2013. 1(43). 211. (in Ukrainian)
- Sprynchuk N.A., Dosenko V.E. Role of determination of mutation D112G in the diagnosis of patients with syndrome of biologically inactive growth hormone among children in Ukraine. Reports of the National Academy of Sciences of Ukraine. Reports of the National Academy of Sciences of Ukraine. 2016. 3. 107-112. http://dspace.nbuv.gov.ua/handle/123456789/99072. (in Ukrainian)