
Международный неврологический журнал Том 18, №3, 2022
Вернуться к номеру
Пілоцитарна астроцитома: огляд літератури
Авторы: Dipak Chaulagain, Volodymyr Smolanka, Andriy Smolanka
Regional Clinical Center of Neurosurgery and Neurology, Uzhhorod National University, Uzhhorod, Ukraine
Рубрики: Неврология
Разделы: Справочник специалиста
Версия для печати
Пілоцитарна астроцитома (ПА) — це часта назва доброякісної пухлини мозку, яка виростає з астроцитів, клітин, що підтримують неврологічну систему. Гарві Кушинг уперше продемонстрував пілоцитарну астроцитому у 1931 році на основі серії астроцитом мозочка. Гліоми, віднесені ВООЗ до I ступеня, мають сприятливий прогноз, і пілоцитарні астроцитоми належать до цієї категорії. У осіб, які брали участь у даному дослідженні, відзначалися симптоми дефіциту функції черепно-мозкових нервів, а також симптоми атаксії та ознаки підвищення внутрішньочерепного тиску. Через зв’язок між розташуванням, розміром пухлини та наявністю супутньої гідроцефалії симптоми та ознаки ПА найчастіше спостерігаються через багато місяців. Найбільш частими симптомами є головний біль, нудота, нечіткість зору, блювання, дискомфорт у спині, підвищення внутрішньочерепного тиску та диплопія. Гістопатологічно ПА має низький або помірний рівень клітинності і складається з клітин з довгими біполярними (волосоподібними) відростками та подовженими і цитологічно м’якими ядрами, а також з ділянок з пухкою, мультиполярною (подібною до протоплазматичних астроцитів) текстурою, що складається з клітин з м’якими округло-овальними ядрами та численними короткими цитоплазматичними розширеннями. Ці ділянки багаті на волокна Розенталя. Найпоширенішим методом лікування ПА є операція з видалення пухлини. Прогноз, як правило, сприятливий, якщо пухлина повністю видалена. Якщо хірургічне втручання неможливе через розташування пухлини, дітям старшого віку та дорослим може бути корисно променеве лікування, щоб допомогти усунути будь-які залишки пухлинних клітин. Іноді використовуються хіміотерапія або інші форми таргетної терапії.
Pilocytic astrocytoma is a frequent name for a benign brain tumor that grows from astrocytes, the cells that support the neurological system. Harvey Cushing showed pilocytic astrocytoma (PA) for the first time in 1931, based on a series of cerebellar astrocytomas. Gliomas classified as grade I by the WHO have a favourable prognosis, and pilocytic astrocytomas fall within this category. There were cranial nerve deficits as well as ataxia symptoms and evidence of elevated intracranial pressure in the individuals in this investigation. Because of the relationship between the tumor’s location, size, and existence of concomitant hydrocephalus, symptoms and signs of PAs are most often seen after many months. The most frequent symptoms are headache, nausea, blurred vision, vomiting, back discomfort, elevated intracranial pressure, and diplopia. Histopathologically PA has a low to moderate level of cellularity and is composed of cells with long bipolar (hair-like) processes and elongated and cytologically bland nuclei, as well as areas with loose, multipolar (protoplasmic astrocyte-like) texture composed of cells with bland, round-to-oval nuclei and numerous short cytoplasmic extensions. These areas are rich in Rosenthal fibers. The most common treatment for PA is surgery to remove the tumor. The prognosis is generally good if the tumor is totally removed. When surgery is not an option because of the tumor’s location, older children and adults may benefit from radiation treatment to help eliminate any leftover tumor cells. Sometimes, chemo or other forms of targeted treatment are used.
астроцитома; пілоцитарна астроцитома; гліома; гліома низького ступеня
astrocytoma; pilocytic astrocytoma; glioma; low grade glioma
1. Introduction
2. Incidence/epidemiology/distribution of pilocytic astrocytoma
/25.jpg)
3. Symptoms/characteristics of pilocytic astrocytoma
4. Histopathology and molecular features of PA
5. Radiological features of PA
6. Adult vs. pediatric age PA
7. Pilocytic astrocytoma vs. рilomyxoid аstrocytoma
8. Pilocytic аstrocytoma vs. рleomorphic xanthoastrocytoma
9. Surgical treatment of adult pilocytic astrocytoma
Conclusions
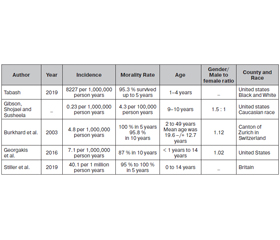
- Yonekawa Y., Lutolf U.M., Kleihues P., Ohgaki H. A population-based study of the incidence and survival rates in patients with pilocytic astrocytoma. J. Neurosurg. 2003. 98. 1170-1174.
- Brown P.D., Buckner J.C., O’Fallon J.R., Iturria N.L., Brown C.A., O’Neill B.P., Scheithauer B.W., Dinapoli R.P., Arusell R.M., Abrams R.A., Curran W.J., Shaw E.G., North Central Cancer Treatment Group, Mayo Clinic. Adult patients with supratentorial pilocytic astrocytomas: a prospective multicenter clinical trial. Int. J. Radiat. Oncol. Biol. Phys. 2004. 58. 1153-1160.
- Ellis J.A., Waziri A., Balmaceda C., Canoll P., Bruce J.N., Sisti M.B. Rapid recurrence and malignant transformation of pilocytic astrocytoma in adult patients. J. Neuro-Oncol. 2009. 95. 377-382.
- Bell D., Chitnavis B.P., Al-Sarraj S., Connor S., Sharr M.M., Gullan R.W. Pilocytic astrocytoma of the adult — clinical features, radiological features and management. Br. J. Neurosurg. 2004. 18. 613-616.
- Gnekow A.K., Kortmann R.D., Pietsch T., Emser A. Low grade chiasmatic-hypothalamic glioma-carboplatin and vincristin chemotherapy effectively defers radiotherapy within a comprehensive treatment strategy — report from the multicenter treatment study for children and adolescents with a low grade glioma — HIT-LGG 1996 — of the Society of Pediatric Oncology and Hematology (GPOH). Klin. Padiatr. 2004. 216. 331-342.
- Bhargava D., Sinha P., Chumas P., Al-Tamimi Y., Shivane A., Chakrabarty A., et al. Occurrence and distribution of pilomyxoid astrocytoma. Br. J. Neurosurg. 2013. 27. 413-8.
- Komotar R.J., Burger P.C., Goldthwaite P.T., Tihan T. Hypothalamic-Chiasmatic Astrocytomas with Pilocytic and Pilomyxoid Morphology: A Reappraisal of 63 Cases. Neurosurgery. 2002. 51. 552-3.
- Komotar R.J., Burger P.C., Carson B.S., Brem H., Olivi A., Goldthwaite P.T., et al. Pilocytic and pilomyxoid hypothalamic/chiasmatic astrocytomas. Neurosurgery. 2004. 54. 72-9.
- Adib S., Schuhmann M., Hempel J., Bornemann A., Zamora R., Tatagiba M. Surgical management of primary and secondary pilocytic astrocytoma of the cerebellopontine angle (in adults and children) and review of the literature. Neurosurgical. Review. 2020. 44(2). 1083-1091.
- Apanisile I., Karosi T. Surgical Management of Pilocytic Astrocytoma of the Optic Nerve: A Case Report and Review of the Literature. Case Reports in Oncological Medicine. 2017. 2017. 1-7.
- Burkhard C., Di Patre P., Schüler D., Schüler G., Yaşargil M., Yonekawa Y., Lütolf U., Kleihues P., Ohgaki H. A population-based study of the incidence and survival rates in patients with pilocytic astrocytoma. Journal of Neurosurgery. 2003. 98(6). 1170-1174.
- Chourmouzi D., Papadopoulou E., Konstantinidis M., Syrris V., Kouskouras K., Haritanti A., Karkavelas G., Drevelegas A. Manifestations of pilocytic astrocytoma: a pictorial review. Insights into Imaging. 2014. 5(3). 387-402.
- Georgakis M., Karalexi M., Kalogirou E., Ryzhov A., Zborovskaya A., Dimitrova N., et al. Incidence, time trends and survival patterns of childhood pilocytic astrocytomas in Southern-Eastern Europe and SEER, US. Journal of Neuro-Oncology. 2016. 131(1). 163-175.
- Kayama T., Tominaga T., Yoshimoto T. Management of pilocytic astrocytoma. Neurosurgical Review. 1996. 19(4). 217-220.
- Chaulagain D., Smolanka V.I., Smolanka A.V., Havryliv T.S. The impact of extent of resection in surgical outcome of pilomyxoid astrocytoma: a case study. Ukrainian Neurosurgical Journal. 2021. 27(4). 43-48. https://doi.org/10.25305/unj.242926.
- Mair M., Wöhrer A., Furtner J., Simonovska A., Kiesel B., Oberndorfer S., et al. Clinical characteristics and prognostic factors of adult patients with pilocytic astrocytoma. Journal of Neuro-Oncology. 2020. 148(1). 187-198.
- Mwita C., Koech F., Sisenda T., Patel K., Macharia B., Rahangdale D. Clinicopathologic Features and Early Surgical Outcome of Astrocytomas in Eldoret, Kenya. Journal of Neurosciences in Rural Practice. 2018. 09(03). 363-369.
- Pompili A., Caperle M., Pace A., Ramazzotti V., Raus L., Jandolo B., Occhipinti E. Quality-of-life assessment in patients who had been surgically treated for cerebellar pilocytic astrocytoma in childhood. Journal of Neurosurgery. 2002. 96(2). 229-234.
- Stiller C., Bayne A., Chakrabarty A., Kenny T., Chumas P. Incidence of childhood CNS tumors in Britain and variation in rates by definition of malignant behaviour: population-based study. BMC Cancer. 2019. 19(1).
- Tabash M. Characteristics, survival and incidence rates and trends of pilocytic astrocytoma in children in the United States; SEER-based analysis. Journal of the Neurological Sciences. 2019. 400. 148-152.
- Theeler B., Ellezam B., Sadighi Z., Mehta V., Tran M., Adesina A., Bruner J., Puduvalli V. Adult pilocytic astrocytomas: clinical features and molecular analysis. Neuro-Oncology. 2014. 16(6). 841-847.
- Voronina N., Aichmüller C., Kolb T., Korshunov A., Ryzhova M., Barnholtz-Sloan J., et al. The age of adult pilocytic astrocytoma cells. Oncogene. 2021. 40(16). 2830-2841.
- Becker A.P, Scapulatempo-Neto C., Neder L., Chimelli L., Reis R.M. Clinical management and evolving novel therapeutic strategies for patients with brain tumors. Intechopen. 2013. 127-143.
- Collins V.P., Jones D.T.W., Giannini C. Pilocytic astrocytoma: pathology, molecular mechanisms and marker. Acta Neuropathol. 2015.
- Salles D., Laviola G., Malinverni A.C., Stávale J.N. Pilocytic astrocytoma: A review of general, clinical, and molecular characteristics. Journal of Child Neurology. 2020. 35(12). 852-858. https://doi.org/10.1177/0883073820937225.
- Koeller K.K., Rushing. E.J. Pilocytic astrocytoma: radiologic-pathologic correlation. Radiographics. 2004. 24(6). 1693-1708.
- Strong J.A., Hatten H.P., Brown M.T., Debatin J.F., Friedman H.S., Oakes W.J., Tien R. Pilocytic Astrocytoma: Correlation between the initial imaging features an clinical aggressiveness. AJR. 1992. 1993(161). 369-372.
- Mubarak F., Naeem A. Imaging and Histopathological Features of Pilocytic Astrocytoma Involving Various Locations of Central Nervous System — Series of Multiple Cases. Journal of Neurophysiology and Neurological Disorders. 2021. 9(101). 1-7.
- Ryu H., Jung T., Lee G., Lee K., Jung S., Jung S., Baek H. Differnces in the clinical couces of pediatric and adult pilocytic astrocytomas with progression: a single-instituition study. Childs Nerv. System. 2015.
- Khan M.A., Godil S.S., Tabani H., Panju S.A., Enam S.A. Clinical review of pediatric pilocytic astrocytomas treated at a tertiary care hospital in Pakistan. Surgicval Neurology International. 2012. 3(90).
- Ding C., Tihan T. Recent progress in the pathology and genetics of pilocytic and pilomyxoid astrocytomas. Balken. Med. J. 2019. 36(2019). 3-11.
- Chaulagain D., Smolanka V., Smolanka A. Diagnosis and management of astrocytoma: a literature review. International Neurological Journal. 2022. 18(1). 23-29. https://doi.org/10.22141/2224-0713.18.1.2022.925.
- Longo M., Adams Perez J., Oliveira F., Antunes A., Vedolin L., Duarte J.A. Pilomyxoid astrocytoma of the corpus callosum presenting with primary haemorrhage in an adolescent. BJR Case Rep. 2010. 2. 20150020.
- Kleinschmidt-DeMasters B.K., Donson Andrew M., Richmond Abby M., Pekmezci Melike, Tihan Tarik, Foreman Nicholas K. SOX10 Distinguishes Pilocytic and Pilomyxoid Astrocytomas From Ependymomas but Shows no Differences in Expression Level in Ependymomas From Infants Versus Older Children or Among Molecular Subgroups. Journal of Neuropathology & Experimental Neurology. 2016. nlw010. doi: 10.1093/jnen/nlw010.
- Ho Chang Y., Supakul Nucharin, Patel Parth U., Seit Vetana, Groswald Michael, Cardinal Jeremy, Lin Chen, Kralik Stephen F. Differentiation of pilocytic and pilomyxoid astrocytomas using dynamic susceptibility contrast perfusion and diffusion weighted imaging. Neuroradiology. 2019. doi: 10.1007/s00234-019-02310-0.