
Газета «Новости медицины и фармации» №11 (781), 2022
Вернуться к номеру
Постковідний синдром: механізми ураження органів-мішеней. Особливості метаболітотропної терапії
Авторы: Кривенко В.І. (1), Колесник М.Ю. (1), Галицька А.К. (2), Кучеренко Л.І. (1, 3), Бєленічев І.Ф. (1, 3), Павлов С.В. (1), Ядловський О.Є. (4)
(1) — Запорізький державний медичний університет, м. Запоріжжя, Україна
(2) — Інститут кардіології ім. акад. М.Д. Стражеска, м. Київ, Україна
(3) — НВО «Фарматрон», м. Запоріжжя, Україна
(4) — Інститут фармакології і токсикології НАМН України, м. Київ, Україна
Разделы: Справочник специалиста
Версия для печати
/11_m.jpg)
У грудні 2019 року в китайському місті Ухань було зареєстровано спалах пневмонії. Спалах був пов’язаний з продовольчим ринком Хуанань. Новий вірус 2019-nCoV було виділено 7 січня 2020 року й ідентифіковано як причину спалаху. Вірус 2019-nCoV швидко поширився по Китаю і багатьом іншим країнам і викликав глобальний спалах, що швидко зростав. 11 лютого 2020 р. ВООЗ назвала хворобу COVID-19 (скорочено від «коронавірусна хвороба 2019»), а 12 березня 2020 р. загальна кількість підтверджених випадків COVID-19 досягла 125 260 у всьому світі, з них 80 981 випадків — у Китаї і 44 279 — за його межами. COVID-19 було оголошено ВООЗ пандемією [1–3]. Станом на 26 травня 2020 р. COVID-19 було підтверджено в 5 404 512 осіб у всьому світі, кількість смертей досягла 343 514 осіб, а рівень смертності становив 6,4 %. Найбільша кількість підтверджених випадків захворювання була в США (1 618 757 випадків) [3, 4].
Клінічні прояви COVID-19
COVID-19 викликає SARS-CoV-2, що належить до підродини бета-коронавірусів. Коронавіруси являють собою оболонкові позитивні одноланцюгові віруси з великою РНК. Хоча перші доступні дані про COVID-19 вказують на можливу передачу вірусу людині від тварин через диких тварин на ринку морепродуктів Хуанань в Ухані [4], епідеміологічні дані й дослідження після цього все частіше демонстрували, що вірус передається від людини до людини повітряно-краплинним шляхом або при прямому контакті, з повідомленнями про те, що в осіб, які не мали прямого контакту з ринком морепродуктів Хуанань, було діагностовано COVID-19, а також про вторинні випадки в лікарнях серед медичних працівників, які мали контакти із пацієнтами з COVID-19. Було підтверджено, що вірус поширюється повітряно-краплинним шляхом при кашлі або чханні [5, 6] із здатністю хазяїна виділяти інфекцію при безсимптомному перебігу [4, 7]. Нині дослідження також передбачають можливу фекально-оральну передачу вірусу [7–9].
Пацієнти з COVID-19 — це переважно дорослі віком понад 18 років, здебільшого чоловіки. Упереджена думка про те, що педіатричні пацієнти не схильні до інфекції, пізніше змінилася у зв’язку з підтвердженими випадками, зареєстрованими в педіатрії в Китаї та всьому світі, проте смертність, як і раніше, є набагато більшою в дорослій групі віком понад 65 років. Дорослі пацієнти із серцево-судинними захворюваннями в анамнезі, респіраторними захворюваннями, ендокринними захворюваннями, діабетом або дорослі з ослабленим імунітетом залишаються найбільш схильними до серйозних ускладнень COVID-19 [3, 8]. Хоча в багатьох пацієнтів з COVID-19 хвороба може перебігати безсимптомно, у деяких хворих розвивається пневмонія, а в 10 % випадків потрібна штучна вентиляція легень і госпіталізація у відділення інтенсивної терапії. У пацієнтів зазвичай відзначаються лихоманка, сухий кашель, задишка, головний біль, нездужання, біль у м’язах і кістках. Менш поширені симптоми включають біль у горлі, сплутаність свідомості, продуктивний кашель, кровохаркання, діарею, нудоту й біль у грудях. Прогресування до пневмонії підтверджується рентгенологічними даними й зазвичай відбувається через 1–2 тижні після появи симптомів. Ознаки пневмонії включають зниження насичення крові киснем, порушення газотранспортної функції крові, мультифокальні затемнення або плямисті/сегментарні консолідації на рентгенограмі грудної клітки або комп’ютерній томограмі (КТ). Пацієнти, які госпіталізовані із запізненням або з погіршенням стану, зазвичай страждають від гострого респіраторного дистрес-синдрому (ГРДС), гострої дихальної недостатності, гострої ниркової недостатності й поліорганної недостатності [6, 8].
Результати лабораторних досліджень при COVID-19
Повна картина крові в пацієнтів із COVID-19 зазвичай показує лімфопенію з тотальною лейкопенією або без неї. Кількість лімфоцитів < 1,0 × 109/л пов’язана з тяжкою формою захворювання [5, 7]. Недавнє дослідження показало, що при тяжких випадках COVID-19 зазвичай має місце високе співвідношення нейтрофілів і лімфоцитів (NLR). NLR розраховується на основі звичайної картини крові шляхом поділу абсолютної кількості нейтрофілів на абсолютну кількість лімфоцитів і вказує на загальний запальний статус пацієнта. Збільшення NLR є фактором ризику смертності не тільки при інфекційних захворюваннях, але й при злоякісних новоутвореннях, гострому коронарному синдромі, внутрішньомозковому крововиливі, поліміозиті, дерматоміозиті [3, 5]. Кількість тромбоцитів зазвичай нормальна або незначно знижена. Рівень С-реактивного білка і швидкість осідання еритроцитів зазвичай підвищені, тоді як рівні прокальцитоніну в нормі, а підвищення рівня прокальцитоніну зазвичай вказує на вторинну бактеріальну інфекцію. Підвищення рівня лактатдегідрогенази, феритину, D-димеру й креатинкінази пов’язане з тяжким перебігом захворювання. Підвищення рівня креатиніну або ферментів печінки (АЛТ і АСТ) відбувається у складних випадках, що прогресують до поліорганної недостатності [5].
Цитокіновий профіль і цитокіновий шторм при COVID-19
Людина, яка пережила цитокіновий шторм —
це вже інша людина.
Ванесса Кастеллі, Аннамарія Чиміні,
Клаудіо Феррі
Інфекція COVID-19 супроводжується агресивною запальною реакцією з вивільненням великої кількості прозапальних цитокінів у події, відомій як цитокіновий шторм. Імунна відповідь хазяїна на вірус SARS-CoV-2 є гіперактивною, що призводить до надмірної запальної реакції. Декілька досліджень, які аналізують профілі цитокінів у пацієнтів з COVID-19, показали, що цитокіновий шторм безпосередньо корелює з пошкодженням легень, поліорганною недостатністю й несприятливим прогнозом тяжкого перебігу COVID-19 [2, 8, 10, 11]. Імунна система має тонкий механізм, здатний реагувати на різні патогени. Нормальна противірусна імунна відповідь потребує активації запальних шляхів імунної системи; однак аберантна або перебільшена відповідь імунної системи хазяїна може викликати тяжке захворювання, якщо його не контролювати [9]. Цитокіни є невід’ємною частиною запального процесу. Цитокіни продукуються кількома імунними клітинами, включно з вродженими макрофагами, дендритними клітинами, природними клітинами-кілерами й адаптивними Т- і В-лімфоцитами. Під час вродженої імунної відповіді на вірусну інфекцію рецептори розпізнавання образів (PRR) розпізнають різні молекулярні структури, характерні для вірусу, що вторгнувся. Ці молекулярні структури називаються молекулярними патернами, асоційованими з патогенами (PAMP). Зв’язування PAMPs з PRRs запускає початок запальної відповіді проти інвазивного вірусу, що приводить до активації декількох сигнальних шляхів, а згодом — факторів транскрипції, які індукують експресію генів, відповідальних за продукцію кількох факторів, що беруть участь в імунній відповіді хазяїна на вірус, серед яких є гени, що кодують кілька прозапальних цитокінів. Основними факторами транскрипції, що активуються PRR, є ядерний фактор κB, білок активації 1, фактори відповіді на інтерферон (IFN) 3 і 7. Ці фактори транскрипції індукують експресію генів, що кодують запальні цитокіни, хемокіни й молекули адгезії. Ця послідовність подій приводить до залучення лейкоцитів і білків плазми до вогнища інфекції, де вони виконують різні функції, що служать для боротьби з ініціюючою інфекцією [5–7].
Найбільш важливими прозапальними цитокінами вродженої імунної відповіді є інтерлейкін (IL) 1, TNF-α і IL-6. Тканинні макрофаги, тучні клітини, ендотеліальні й епітеліальні клітини є основним джерелом цих цитокінів під час уродженої імунної відповіді. Цитокіновий шторм виникає внаслідок раптового гострого збільшення циркулюючих рівнів різних прозапальних цитокінів, включно з IL-6, IL-1, TNF-α та інтерфероном. Це збільшення цитокінів призводить до притоку різних імунних клітин, таких як макрофаги, нейтрофіли й Т-клітини, з кровотоку у вогнище інфекції з деструктивною дією на тканини людини внаслідок дестабілізації міжклітинних взаємодій ендотеліальних клітин, пошкодження судинного бар’єра, капілярів. Надалі це може призводити до дифузного альвеолярного ушкодження, розвитку поліорганної недостатності і зрештою — до смерті [10]. Гострий респіраторний дистрес-синдром, що призводить до низького рівня насичення киснем, є основною причиною смерті при COVID-19. Хоча точний механізм ГРДС у пацієнтів з COVID-19 до кінця не зрозумілий, надлишкове вироблення прозапальних цитокінів вважається одним з основних факторів, що йому сприяють [5, 8, 10, 11].
Накопичені дані свідчать, що деякі пацієнти з тяжкою формою COVID-19 страждають від цитокінового шторму. Аналіз рівнів цитокінів у плазмі 41 підтвердженого випадку COVID-19 у Китаї виявив підвищені рівні IL-1β, IL-7, IL-8, IL-9, IL-10, FGF, G-CSF, GM-CSF, IFN-γ, IP-10, MCP-1, MIP-1A, MIP1-B, PDGF, TNF-α і васкулоендотеліального фактора росту (VEGF) як у пацієнтів, що надійшли у відділення інтенсивної терапії, так і в пацієнтів, які не перебувають у відділенні інтенсивної терапії, порівняно зі здоровими дорослими. У всіх пацієнтів, включених у дослідження, була пневмонія, 1/3 пацієнтів було госпіталізовано до відділення інтенсивної терапії, і шість із цих пацієнтів померли [7, 9].
Багатоцентрове ретроспективне дослідження 150 пацієнтів із COVID-19 у Китаї оцінювало предиктори смертності від COVID-19. У дослідженні проаналізовано дані 82 випадків захворювання в пацієнтів, які вилікувалися від COVID-19, і 68 випадків смерті від COVID-19, і було повідомлено про значно вищі рівні IL-6 у випадках смерті, ніж у випадках одужання. В іншому дослідженні, що аналізує дані 21 пацієнта в Китаї, повідомлялося про підвищені рівні IL-10, IL-6 і TNF-α у тяжких випадках (n = 11) порівняно з випадками середньої тяжкості (n = 10) [7]. В аналогічному дослідженні Gao та ін. оцінили 43 пацієнтів в Китаї і повідомили, що рівні IL-6 були значно вищими в тяжких випадках (n = 15), ніж у легких випадках (n = 28) [22]. Так само Чен і співавт. вивчили загалом 29 пацієнтів з COVID-19, розділених на три групи відповідно до діагностичних критеріїв, і виявили, що ІL-6 був вищим у критичних випадках (n = 5), ніж у тяжких випадках (n = 9), і що IL-6 був вищим у тяжких випадках, ніж у легких (n = 15) [11, 12–15].
Поки немає достатньої кількості даних про тяжких педіатричних пацієнтів із COVID-19. Дослідження, у якому оцінювалися вісім тяжкохворих китайських педіатричних пацієнтів з COVID-19, які отримували лікування у відділенні інтенсивної терапії, віком від 2 місяців до 15 років, показало підвищені рівні IL-6, IL-10 і IFN-γ серед інших лабораторних результатів [10].
Цитокіновий шторм — критичний життєпогрозливий стан, що вимагає госпіталізації в реанімацію і має досить високу летальність. Цитокіновий шторм характеризується клінічними проявами вираженого системного запалення, гіперферитинемії, гемодинамічної нестабільності й поліорганної недостатності й за відсутності лікування призводить до смерті. Тригером для цитокінового шторму є неконтрольована імунна відповідь, що призводить до постійної активації та розширення імунних клітин, лімфоцитів і макрофагів, які виробляють величезну кількість цитокінів, що призводить до цитокінового шторму. Клінічні ознаки цитокінового шторму пов’язані з дією прозапальних цитокінів, таких як IL-1, IL-6, IL-18, IFN -γ і TNF-α [11].
Цитокіновий шторм був зареєстрований при кількох вірусних інфекціях, включно з тими, що викликані вірусом грипу H5N1 [12, 13], вірусом грипу H1N1 і двома коронавірусами, тісно пов’язаними з COVID-19: ТОРС-КоВ і БВРС-КоВ [5, 13]. Як прозапальні цитокіни (наприклад, IL-1, IL-6 і TNF-α), так і протизапальні цитокіни (наприклад, IL-10 та антагоніст рецептора IL-1) підвищені в сироватці пацієнтів із цитокіновим штормом. Основними учасниками взаємодії цитокінового шторму є IL-6 і TNF-α. За відсутності негайного й адекватного терапевтичного втручання в пацієнтів розвивається гострий дистрес-синдром унаслідок гострого ураження легень з подальшою поліорганною недостатністю й летальним кінцем. Отже, цитокіновий шторм вимагає негайного медикаментозного втручання, інакше це може призвести до смерті. Припускається, що крім противірусної терапії, яка може безпосередньо впливати на вірус, також і протизапальна терапія, що зменшує реакцію цито–кінів, знижує як захворюваність, так і смертність у пацієнтів з COVID-19.
Раннє розпізнавання цитокінового шторму й своєчасне лікування можуть привести до кращого результату терапії. Для лікування цитокінового шторму було запропоновано кілька біологічних агентів, спрямованих на цитокіни. Було доведено, що антагоніст рецептора IL-1, анакінра, а також препарат РАІЛ, який використовується при лікуванні ревматоїдного артриту, допомагають при цитофагічному гістіоцитарному панікуліті з вторинним гемофагоцитарним лімфогістіоцитозом, при хронічної ішемії органів і тканин, що пов’язана з тяжким цитокіновим штормом [8, 9]. Тоцилізумаб є рекомбінантним препаратом антагоніста рецептора IL-6, який перешкоджає зв’язуванню IL-6 з його рецептором і блокує передачу сигналу. Тоцилізумаб використовується для лікування ревматоїдного артриту, ювенільного ідіопатичного артриту, гігантоклітинного артеріїту; також він довів свою ефективність у лікуванні КС, викликаного терапією CAR-T-клітинами при гематологічних злоякісних новоутвореннях [12]. Інгібітори цитокінів, наприклад інгібітори JAK, також вивчаються для корекції цитокінового шторму.
Цитокіновий шторм, мабуть, є однією з найчастіших причин смертності під час нещодавно оголошеної пандемії COVID-19. Терапевтичні підходи до управління цитокіновим штормом COVID-19 можуть забезпечити можливість зниження захворюваності й смертності, пов’язаних із COVID-19, і будуть перебувати в центрі уваги в майбутніх дослідженнях.
Оскільки IL-6 є цитокіном, рівень якого найбільше підвищений у пацієнтів з COVID-19 і безпосередньо пов’язаний з більш високою смертністю, тоцилізумаб є препаратом-кандидатом для використання в лікуванні цитокінового шторму. Обнадійливі результати були отримані в Китаї, де тоцилізумаб застосовувався для лікування 1027 пацієнтів з тяжким і критичним перебігом COVID-19. Клінічні дані показали, що симптоми, гіпоксемія і зміни легень на КТ зменшувалися відразу після лікування тоцилізумабом у більшості пацієнтів. Усе це обґрунтовує застосування тоцилізумабу для лікування цитокінового шторму, пов’язаного з тяжкими формами COVID-19. У США FDA схвалило клінічне дослідження ІІІ фази клінічних випробувань тоцилізумабу в госпіталізованих пацієнтів з тяжкою пневмонією, спричиненою COVID-19.
Ендотеліальна дисфункція при COVID-19
Фізіологічні функції ендотелію включають контроль судинного тонусу, тканинного гемо–стазу, цілісності бар’єра, запалення, окиснювального стресу, судинної проникності й структурної та функціональної цілісності. Низка видів вірусів, таких як лихоманка денге, лихоманка Ебола й цитомегаловірус, можуть інфікувати ендотеліальні клітини й викликати ендотеліальну дисфункцію [12–15]. На сьогодні дослідники отримують дедалі більше даних, які проливають світло на формування ендотеліальної дисфункції — важливої ланки патогенезу COVID-19 [11, 12]. Гістопатологічні спостереження показали, що COVID-19 призводить до розвитку ендотеліальної дисфункції, у формуванні якої цитокіновий шторм і оксидативний стрес відіграють фундаментальну роль. Після зараження SARS-CoV-2 і виявлення ендотеліопатії в пацієнтів виявлено зниження експресії ендо–теліальної синтази монооксиду азоту (eNOS), зниження біодоступності NO, а на фоні підвищення індуцибельної синтази монооксиду азоту (iNOS) — активацію нітрозативного стресу. NO також має антитромботичну дію, запобігає адгезії лейкоцитів і тромбоцитів до активованого ендотелію, тим самим інгібуючи тромбоз і розвиток атеросклеротичних бляшок.
Загальновідомо, що NO є нестабільним, короткоживучим радикалом і для його стабілізації і подальшого транспортування передбачені такі механізми, як утворення з низькомолекулярними тіолвмісними сполуками (глутатіон, цистеїн, метіонін) стійких S-нітрозольних комплексів [13]. В умовах дефіциту тіольних сполук (оксидативний стрес, ішемія, інтоксикації, гіпертонічна хвороба тощо) порушується транспорт NO, тому що він піддається атаці таких активних форм кисню (АФК), як супероксидрадикал і гідроксилрадикал, із перетворенням на цитотоксичний продукт — пероксинітрит, про що свідчить підвищення специфічного маркера пероксинітриту — нітротирозину. При цьому спостерігається інтенсифікація оксидативного й нітрозативного стресу. Інтенсифікація нітрозативного стресу спостерігається на тлі порушень у тіол-дисульфідній системі — зниження рівня відновлених тіолів і підвищення рівня його окиснених інтермедіатів на тлі пригнічення активності глутатіон-залежних ферментів (глутатіонредуктази, глутатіонпероксидази й глутатіон-S-трансферази). Зниження біодоступності NO частково відбувається через зниження продукції NO у системі eNOS — L-аргінін за рахунок гіперпродукції виробництва АФК, які інактивують eNOS і гальмують експресію мРНК eNOS. При формуванні дисфункції ендотелію при COVID-19 важливе значення має експресія ферментів, що регулюють рівень АФК — супероксиддисмутази й каталази.
Нашими дослідженнями було виявлено пригнічення активності каталази й Мп-СОД. Гострий респіраторний дистрес-синдром є найбільш загрозливим механізмом гострого пошкодження легень, а також таким, що зумовлює летальність, пов’язану з COVID-19. Ендотелій легеневих капілярів готує ґрунт для проникнення вірусу, реплікації, тим самим полегшуючи проникнення вірусу в циркулюючу кров [12, 13]. SARS-CoV-2 інфікує ендотеліальні клітини й епітеліальні клітини в тканинах легень через ангіотензинперетворюючий фермент 2 (АПФ-2) і відповідні рецептори на клітинах-хазяях [14]. Цікаво, що тільки живий вірус SARS-CoV-2 викликає підвищену проникність ендотелію за рахунок інфільтрації прозапальних клітин до периваскулярної тканини, а також інтерстиціального набряку й затримки рідини в альвеолярних просторах. Крім того, вивільнення прозапальних цитокінів у період після викликаного SARS-CoV-2 цитокінового шторму призводить до порушення біологічних бар’єрів, легеневої гіпертензії і фіброзу легень.
У даний час АПФ-2 описують як перший ідентифікований рецептор, відповідальний за проникнення SARS-CoV-2 у клітину-хазяїна включно з ендотеліоцитами. Аналіз експресії АПФ-2 у тканинах, взятих для автопсії, показує, що його значна експресія спостерігається переважно в дрібних судинах і капілярах і менше виражена в артеріолах/венах у пацієнтів з COVID-19. Дані про експресію АПФ-2 у коронарних судинах суперечливі, хоча ступінь тяжкості COVID-19 безпосередньо залежить від ураження серця. У нормі ендотеліальні клітини легень практично не експресують АПФ-2, а при розтині пацієнтів, які померли від COVID-19, у судинах легень виявлено збільшення експресії АПФ-2 у кілька разів. З віком експресія АПФ-2 у легеневих ендотеліоцитах прогресує з можливою участю IL-7 за рахунок NF-κB-залежного механізму. CD209L/L-SIGN був ідентифікований як ще один рецептор, що опосередковує проникнення SARS-CoV-2 у клітину людини, який також може взаємодіяти з АПФ-2 для полегшення проникнення SARS-CoV-2. L-SIGN взаємодіють з N-гліканами на рецепторно-зв’язувальному домені зв’язуючого білка SARS-CoV-2 за участю Ca2+. Електронна мікроскопія показує коронавіруси й везикули, що містять частинки віріону, у венозних ЕК з високою експресією АПФ-2.
Зміна під дією вірусу рецепторно-зв’язуючого домену шипоподібного білка S1 (S1-RBD) в ендотеліальних клітинах мікросудин головного мозку індукує деградацію ендотеліальних з’єднувальних білків (VE-кадгерин, сполучна молекула адгезії A, коннексин-43 і PECAM-1), тим самим порушуючи ендотеліальну бар’єрну функцію, підвищуючи проникність судин і посилюючи формування дисфункції ендотелію в пацієнтів із COVID-19. Ці дані свідчать про діагностичну цінність шипоподібних білків з метою прогнозу дисфункції ендотелію при COVID-19, а також можливості їх розгляду як перспективної мішені фармакокорекції.
Крім шипоподібного білка, нуклеокапсидний білок SARS-CoV-2 також може сприяти ініціюванню механізмів пошкодження ендотелію через сигнальні шляхи TLR2/NF-κB і MAPK. Недавнє дослідження показало, що рівень маркерів запалення (IL-6, TNF-α, ICAM-1 і каспаза-1, рецептор протеїнкінази С) значно підвищувався в ендотеліальних клітинах пацієнтів із легкою формою COVID-19. Крім того, було продемонстровано, що в пацієнтів із COVID-19 спостерігається підвищення експресії інфламасоми NLRP3, IL-1β, iNOS і дефіцит антиапоптичного білка bcl-2 в ендотеліальних клітинах легень. Дані РНК-секвенування показують підвищену експресію маркерів активації ендотелію, таких як RELB (субодиниця p50) і TNF-α в ендотеліоцитах пацієнтів з легкою формою COVID-19. Отже, гіперзапалення й активація запальних процесів, пов’язаних з інфекцією SARS-CoV-2, призводить до ураження й ініціювання програмованої смерті ендотеліальних клітин (наприклад, до піроптозу).
Глікокалікс являє собою багату на протеоглікани й глікопротеїни мікроструктуру, що покриває ендотелій, необхідну для судинного гомеостазу за допомогою регуляції судинного тонусу, проникності, тромбозу й адгезії лейкоцитів до ендотелію. Глікокалікс складається із сильно сульфатованих протеогліканів з бічними ланцюгами глікозаміногліканів. Неушкоджена бар’єрна структура сульфатованого глікокаліксу ендотелію може відштовхувати SARS-CoV-2. Порушена структура глікокаліксу призводить до гіперзапальної реакції й окиснювального стресу, що спричиняє підвищену чутливість до інфекції SARS-CoV-2. Нещодавнє дослідження з використанням методу мас-спектроскопії виявило зв’язування глікокаліксу з білком Spike, що пов’язане із взаємодією S-білка/AПФ-2. Повідомлялося, що в пацієнтів з COVID-19 спостерігалося підвищення рівня ендотеліального глікокаліксу й експресії синдекану-1 (SDC-1). Навіть у пацієнтів з COVID-19, які одужують, рівень SDC-1 був значно підвищений порівняно зі здоровими людьми, що свідчить про наявність стійкого ендотеліального пошкодження після тяжкого прогресування COVID-19. Синдекан-1, необхідний судинний компонент глікокаліксу, що вивільняється після васкуліту й ускладнень, добре корелює, зокрема, з маркером згортання крові (D-димером). Він являє собою потенційний біомаркер для моніторингу прогресування серцево-судинних ускладнень при COVID-19 [12–15]. Рівень синдекану-1 у хворих корелює з рівнем тромбомодуліну, TNF-α і IL-1b, IL-6 і вираженістю порушень еластичності судин (інструментальні методи дослідження). Білковий компонент глікокаліксу може бути розщеплений відповідним ферментом, таким як гепариназа. У плазмі, отриманій від пацієнтів із COVID-19, спостерігалися підвищені активність гепаранази і рівень гепарансульфату. У пацієнтів з COVID-19 відбувається відщеплення глікокаліксу й руйнування ендотеліальних клітин, чому можна запобігти за допомогою гепарину або низькомолекулярних гепариноїдів. Ці результати показують, що здоровий стан глікокаліксу має виняткове значення для виявлення судинного гомеостазу й запобігання зв’язуванню вірусу. Засоби для захисту або відновлення вмісту ендотеліального глікокаліксу мають великий терапевтичний потенціал у комплексному лікуванні COVID-19.
Ретроспективне дослідження показало, що рівні розчинних ICAM-1, VCAM-1 і білків судинної адгезії-1 (VAP-1) підвищені в пацієнтів з COVID-19 і змінюються під час прогресування й регресії захворювань. Ці дані обґрунтовують використання цих молекулярних маркерів у прогностичному аналізі дисфункції ендотелію при COVID-19. Як діагностичний маркер розглядається резистин, що являє собою пептидний гормон, секретований жировою тканиною, і бере участь у запальній реакції в ендотелії судин. Рівень резистину в плазмі підвищений у пацієнтів з COVID-19 і пов’язаний з тяжкістю захворювання, а також з експресією запальних цитокінів (IL-6, IL-8 і MCP-1) і молекулярних адгезій (ICAM1 і VCAM1). COVID-19 може призвести до активації ангіогенезу, про що свідчить підвищення в крові пацієнтів рівня васкулоендотеліального фактора — VEGF-A.
У пацієнтів з COVID-19 підвищена секреція множинних маркерів активації/дисфункції ендотелію, таких як D-димер (маркер коагулопатії і системного тромбозу), vWF (основний компонент шляху згортання крові й медіатор судинного запалення) і молекули запалення, що виділяються з тілець Вейбеля — Паладе), фактор VIII (маркер коагуляції), PAI-1 (маркер пошкодження й старіння ендотелію), розчинний тромбомодулін (рТМ), розчинний Р-селектин (маркер тромбоцитарної та ендотеліальної активації), розчинний ICAM1 (sICAM1, маркер ендотеліального запалення), розчинний VCAM1 (sVCAM1, маркер ендотеліального запалення), ангіопоетин-2 (Ang-2, маркер ангіогенезу й тромбозу), розчинний Е-селектин (sE-селектин, маркер ендотеліального запалення), ET1 (потужний вазоконстриктор) [15, 16].
Крім того, рівні ендотеліальних маркерів були підвищені в пацієнтів з COVID-19, які не вижили, порівняно з тими, хто вижив. Рівні біомаркерів активації/пошкодження ендотеліальних клітин добре корелюють з підвищенням експресії прозапальних цитокінів і хемокінів. Ці дані мають потенційну прогностичну цінність для оцінки ступеня тяжкості й летальності при COVID-19. У пацієнтів з COVID-19 спостерігається кореляція рівня молекулярних маркерів зі зниженням ацетилхолін-опосередкованої дилатації судин (легкодоступний метод оцінки ендотеліальної дисфункції), що підтверджує їх інформаційну цінність. Комбінація кількох маркерів може мати виняткову цінність для діагностики ендотеліальної дисфункції, коагулопатії та тромбоемболії, а також дозволяє прогнозувати госпіталізацію пацієнтів із COVID-19 у відділення інтенсивної терапії.
Отже, нові методи лікування, спрямовані на зменшення ендотеліальної дисфункції, і включення до комплексної терапії постковідного синдрому ендотеліопротекторів можуть значно поліпшити якість лікування COVID-19.
Пошкодження міокарда при COVID-19
Хоча COVID-19 у першу чергу пошкоджує легені, він може негативно впливати на інші органи, зокрема на серце. COVID-19 значно підвищує смертність пацієнтів від ускладнень з боку серцево-судинної системи, зокрема артеріальної гіпертензії. Найбільш поширені серцево-судинні ускладнення при COVID-19 — аритмія, ішемія міокарда (у пацієнтів підвищений рівень тропоніну I, МВ-креатинфосфокінази, NT-proBN, білка ST2), коагуляція (про що свідчить підвищений рівень D-димеру), фульмінантний міокардит, серцева недостатність та ініціація молекулярних механізмів атеросклерозу [14, 15]. Предиктором цих серцево-судинних ускладнень є ендотеліальна дисфункція. Також відомо, що пацієнти з підвищеною експресією гена й білка АПФ-2 переносять COVID-19 у тяжкій формі і серед них більший відсоток летальних наслідків від серцево-судинних ускладнень [2]. Крім того, COVID-19 є важливим фактором ризику розвитку гострого інфаркту міокарда [15]. Дані багатоцентрового реєстру свідчать про те, що в пацієнтів з інфарктом міокарда з підйомом сегмента ST, зареєстрованих під час перших хвиль COVID-19, спостерігається більш виражене ішемічне пошкодження міокарда (за даними біохімічних і молекулярних досліджень та електрокардіограми (ЕКГ)) і більша частота смертельних випадків. Тому багато клініцистів наполягали на необхідності вакцинації проти COVID-19 у пацієнтів цих груп. Однак дані невеликої дослідницької групи свідчать про те, що в більшості пацієнтів з тяжким інфарктом міокарда симптоми розвинулися після щеплення від COVID-19.
Імунітет
SARS-CoV-2 на 73 % гомологічний SARS-CoV, і, як і у випадку з SARS-CoV, патогенез пневмонії, спричиненої SARS-CoV-2, має дві фази. Вірусна фаза характеризується реплікацією вірусу, що призводить до прямого ураження тканин вірусом. Ступінь цього ушкодження визначає патогенез вторинної фази, яка характеризується рекрутуванням ефекторних імунних клітин, що викликають місцеву й системну запальну реакцію, яка може зберігатися навіть після елімінації вірусу. Нездатність забезпечити своєчасну й ефективну імунну відповідь проти SARS-CoV-2 є багатофакторною і пояснюється дефектами у відповіді IFN типу I.
Своєчасна продукція IFN типу I клітинами-хазяями має вирішальне значення для обмеження реплікації вірусу й підвищення противірусного імунітету. Варіанти втрати функції в локусах, які контролюють TLR3- і IRF7-залежний імунітет до IFN I типу, були ідентифіковані в невеликої кількості тяжких пацієнтів [1–3, 18, 19]. Слід зазначити, що це пацієнти старшої вікової групи (50–70 років), які до захворювання на COVID-19 ніколи не були госпіталізовані з приводу тяжкого вірусного захворювання. Автоантитіла проти IFN-α і IFN-ω були ідентифіковані в пацієнтів з тяжким захворюванням, і було показано, що вони сприяють уповільненню елімінації вірусу. Нещодавно було виявлено, що нейтралізуючі IFN-α і IFN-ω автоантитіла підвищуються після 50–60 років. Автоантитіла до IFN I типу можуть передувати захворюванню й можуть використовуватися як біомаркери ступеня тяжкості COVID-19. Незбалансований адаптивний імунітет на тлі тяжкої Т-клітинної лімфопенії, спричиненої секвестрацією Т-клітин у тканинах або апоптозом Т-клітин внаслідок прозапальних цитокінів, часто зустрічається в тяжких пацієнтів. Було показано, що дефекти імунної відповіді типу 1 і надлишковий імунітет типу 2 корелюють з тяжким перебігом COVID-19, що дозволяє припустити, що неадаптована адаптивна імунна відповідь на вірус також може призвести до затримки елімінації вірусу й прогресування захворювання. Значна експансія плазмобластів, що досягає 30 % циркулюючих В-клітин, яка іноді пов’язана з екстрафолікулярними реакціями, також була зареєстрована в тяжких пацієнтів. Масивна експансія плазмобластів може відображати поліреактивність з огляду на низькі рівні соматичних мутацій у клонах антитіл, що спостерігаються в пацієнтів, і може мати менш потужний вірусний контроль, сприяючи пошкодженню тканин.
Розтин померлих пацієнтів із COVID-19 виявив значне накопичення активованих імунних клітин, що дозволяє припустити, що ураження органів-мішеней викликане не лише вірусом, але й надмірною активацією імунної системи. Так, поява надлишку циркулюючих незрілих моноцитів, нейтрофілів і мієлоїдних клітин-попередників, що називають екстреним мієлопоезом, є майже патогномонічною для тяжкого захворювання, і цей процес запускається під час першої фази інфекції, імовірно, через уповільнений кліренс вірусу, особливо в умовах уже існуючого зміненого мієлопоезу. Циркулюючі мієлоїдні клітини продукують надмірну кількість запальних молекул, які сприяють проникності судин і пошкодженню органів. У тяжких пацієнтів спостерігається дефіцит макрофагів у легеневій тканині, які відіграють ключову роль у тканинному гомеостазі й відновленні. Механізм дефіциту легеневих макрофагів до кінця не з’ясований — це може бути результатом прямого вірусного пошкодження або результатом загибелі клітин, викликаної надмірним запаленням. При тяжкому перебігу COVID-19 були виявлені автоантитіла, зокрема велика частка антитіл, спрямованих на ядерний антиген, фосфоліпіди, антигени Т-клітин, антигени В-клітин, хемокіни й цитокіни. Антифосфоліпідні антитіла викликають утворення тромбів в експериментах на мишах, тоді як імуноглобулін G (IgG) від пацієнтів з анти-CD38 автоантитілами виявляє підвищений клітинний фагоцитоз макрофагами, що може сприяти вираженій лімфопенії. Автоантитіла також були виявлені в спинномозковій рідині пацієнтів з неврологічними порушеннями в постковідний період.
Крім того, афукозильовані антитіла IgG1 проти SARS-CoV-2 накопичуються переважно в пацієнтів чоловічої статі з тяжкою формою COVID-19. Ці антитіла стимулюють вироблення запальних цитокінів і дегрануляцію NK-клітин, що може сприяти пошкодженню тканин головного мозку, міокарда, ендотелію судин. Ці антитіла відсутні в безсимптомних пацієнтів і серопозитивних дітей. Механізми, які призводять до продукції афукозильованих антитіл, а також питання, чи можуть ці антитіла індукуватись при вакцинації, до кінця не з’ясовано.
Неврологічні порушення при COVID-19
Приблизно в 30 % пацієнтів із COVID-19 спостерігалися неврологічні симптоми з проявами від легких до тяжких, у тому числі головний біль, запаморочення, порушення свідомості, енцефалопатія, аносмія, гіпогевзія та гіпосмія. Нейротропізм вірусу SARS-CoV-2 пояснює його нейроінвазію, що провокує неврологічні пошкодження, такі як гостра демієлінізація, нейрозапалення тощо. На молекулярному рівні в пацієнтів з COVID-19 були вищі рівні цитокінів і хемокінів (цитокіновий шторм), які порушують проникність гематоенцефалічного бар’єра (ГЕБ), що дозволяє моноцитам, лімфоцитам, продуктам ліпопероксидації, окисної модифікації білка, продуктам нітрозилювання білків і ДНК проникати в головний мозок, викликаючи нейрозапалення, нейродегенерацію і демієлінізацію.
Серед механізмів вторинного пошкодження головного мозку особливе значення мають реакції локального запалення навколо зони ядра інфаркту, що супроводжуються різким підйомом рівня запальних цитокінів, зокрема IL-1b [20, 21]. При взаємодії IL-1b з рецепторами активуються фактори ядерної транскрипції AP-1 і NF-κB, які змінюють поведінку клітин-мішеней і призводять до розвитку гострофазної клітинної відповіді, експресії інших прозапальних факторів, стимуляції астроцитами експресії iNOS і цитотоксичних похідних NO, підвищення проникності мітохондріальної пори й ініціації нейроапоптозу [21]. Виявлено кореляційний взаємозв’язок між підвищенням рівня прозапальних цитокінів (IL-1b, TNF-α, IL-6, IL-8), ступенем тяжкості неврологічних порушень і дефіцитом ростових факторів (BDNF, IGF-1, PDGF). Сигнальний шлях з IL-1b, спрямований на посилення відстрочених механізмів загибелі нейронів після COVID-19, може регулюватися HSP70 [13, 19, 22]. Підвищення концентрації HSP70 у межах фізіологічної норми приводить до підвищення IL-1β до рівня, необхідного для участі в цито- і нейропротекції, дефіцит HSP70 може спричинити значну експресію IL-1. Надекспресія HSP70 послаблює експресію IL-1β за рахунок інгібування факторів транскрипції C/EBPβ і C/EBPδ [13, 22, 23]. HSP70 може запобігати продукції запальних цитокінів шляхом втручання в NF-κB-залежну транскрипцію [17, 18].
Відомо, що підвищена продукція TNF-α, IL-1β, IL-6 та IL-8 відбувається на тлі дефіциту відновленого глутатіону [11, 12]. Відновлений глутатіон і його попередник N-ацетилцистеїн здатні модулювати NF-κB, знижувати експресію IL-1β і виявляти протизапальну дію [17, 19]. IL-1β залежно від концентрації може регулювати транспорт відновленого глутатіону й впливати на механізми нейропротекції/нейродеструкції [20]. Інтермедіат тіол-дисульфідної системи глутатіон є важливим компонентом захисту нейрона, підвищує його стійкість до гіпоксії, обмежує гіперзбудливість NMDA, виступає як резерв цистеїну в клітині, регулює синтез і стабільність HSP70, гальмує NO- та IL-1b-залежні механізми.
Виявлено, що депривація рівня відновленого глутатіону в нейронах призводить до падіння HSP70, виявлено кореляційний взаємозв’язок між вираженістю неврологічних порушень і дефіцитом відновленого глутатіону й HSP70 при церебральній ішемії, нейроінфекції, нейродегенеративних захворюваннях [2, 3, 19]. Раціональна нейропротекція передбачає переривання каскадних механізмів загибелі нейронів, що і визначає IL-1b як важливу мішень фармакологічного впливу для запобігання неврологічним ушкодженням, викликаним інфекцією SARS-CoV-2, або їх зменшення [20].
Активація нейроапоптозу, на думку багатьох дослідників, є першопричиною розвитку стійких порушень когнітивно-мнестичних функцій центральної нервової системи (ЦНС), викликаних інфекцією SARS-CoV-2. Нейроапоптоз розвивається як каскадний процес, що супроводжується активацією (індукцією утворення) специфічних про- або антиапоптичних білків, а також спеціальних протеолітичних ферментів — каспаз. Серед факторів запуску апоптозу слід відзначити утворення активних форм кисню в процесі «збоченого» шляху окисного метаболізму в клітині. Існують переконливі докази того, що центральна роль у продукції АФК і подальшому розвитку апоптозу і некрозу належить мітохондріям, зміні проникності їх мембран у результаті формування специфічного комплексу мітохондріальних пор та ініціювання мітоптозу. У цьому зв’язку перспективним є застосування засобів, які виявляють мітопротективну дію (нейротрофічні церебропротектори, антиоксиданти, коензими). Як перспективні мішені нейропротекції при COVID-19 розглядаються гістондеацетилази (HDACi) й застосування інгібіторів гістондеацетилази (HDACi) через їх нейропротекторну дію. Так, інгібітори гістонацетилази здатні гальмувати нейроапоптоз, регулювати експресію генів нейротрофічного фактора головного мозку (BDNF) і нейротрофічного фактора глії (GDNF), а також генів білків — транспортерів зворотного захоплення серотоніну і генів білків пам’яті [21, 23].
У зв’язку з цим досліджуються відомі лікарські препарати, у механізмі дії яких є ланка, спрямована на інгібування гістонацетилази. Це вальпроат натрію (депакін), оксибутират натрію, фенілбутират натрію (лікарський засіб для лікування порушення циклу сечовини). Крім того, інгібітори гістонацетилази пригнічують експресію прозапальних цитокінів (IL-6, IL-1b і TNF-α), знижуючи нейротоксичність, що також обґрунтовує привабливість гістонацетилази як перспективної мішені нейропротекції. Інгібітори гістонацетилази також допоможуть уникнути проникнення вірусу в центральну нервову систему і зменшити реплікацію вірусу за рахунок пригнічення рецепторів вірусу. Клінічними спостереженнями протягом 2019–2022 років встановлено, що понад 15 % пацієнтів страждають від інсульту внаслідок COVID-19. Дослідження показали, що COVID-19 запускає хвилю запальних цитокінів, які викликають дисфункцію ендотеліальних клітин і коагулопатію, яка збільшує ризик інсульту або тромбозів [19–22].
Запалення ендотелію після інфекції може також дестабілізувати атеросклеротичну бляшку й спричинити тромботичний інсульт. Також були повідомлення про геморагічний інсульт, пов’язаний з COVID-19, чому сприяє коагулопатія, про що свідчить значне підвищення протромбінового часу в постковідних пацієнтів. Також фактором, що сприяє геморагічному інсульту, є підвищення артеріального тиску, спричинене інфекцією, що призводить до зниження рівня АПФ-2. Усе це призводить до дисбалансу ренін-ангіотензинової системи, зрештою, до ендотеліальної дисфункції, вазоконстрикції і порушення кровообігу в життєво важливих органах [11, 15, 17, 21].
Інші неврологічні порушення, пов’язані з COVID-19, включають енцефалопатію, аносмію, енцефаліт, психоз, сплутаність свідомості, головний біль, депресію і тривогу. Хоча в літературі повідомляється про кілька гіпотез, єдиний патофізіологічний механізм цих розладів залишається незрозумілим. Легенева дисфункція, що призводить до поганої оксигенації мозку, може пояснити енцефалопатію та інші розлади в пацієнтів із COVID-19. Вважають, що причиною цих станів може бути пряме ураження вірусом центральної нервової системи або порушення гематоенцефалічного бар’єра прозапальними цитокінами, рівень яких значно зростає під час COVID-19. Проте взаємозв’язок між рівнями запальних цитокінів і такими станами, як депресія та тривога, суперечливий. Можливо, соціальна ізоляція під час пандемії частково може бути фактором, що сприяє депресії.
Відомо, що COVID-19 призводить до дисбалансу серотонінових рецепторів 5НТ1А/5НТ2А, підвищення чутливості останніх. Як відомо, гіперзбудливість 5НТ2А призводить до дезорієнтації, деперсоналізації, ажитації, галюцинацій, суїциду, а десенситизація 5НТ1А — до депресій, і особливо депресій, резистентних до антидепресантів. Нещодавні дослідження причин депресії після COVID-19 виявили, що навіть після послідовного лікування кількома антидепресантами майже 35 % пацієнтів не досягли ремісії. Передбачається, що в цих депресіях може бути епігенетичний компонент, який не усувається сучасними фармакологічними методами лікування. У пацієнтів після COVID-19 у середині депресивного епізоду виявили підвищену експресію мРНК гістонацетилази С2 і С5 порівняно з контрольною групою і пацієнтами в стадії ремісії. Гістонацетилази можуть регулювати експресію білків — транспортерів зворотного захоплення трансмітерів [22].
Є дані клінічних спостережень про поширеність неврологічних симптомів серед 2533 госпіталізованих пацієнтів з COVID-19. Неврологічні симптоми спостерігалися в 73 % госпіталізованих пацієнтів з COVID-19 і переважно включали головний біль, міалгії і порушення свідомості. Ураження центральної нервової системи, про які повідомлялося при COVID-19, були переважно неспецифічними енцефалопатіями, які становили від 13 до 40 % усіх неврологічних порушень. Це постінфекційні синдроми, що включають гострий демієлінізуючий енцефаломієліт, гостру некротизуючу енцефалопатію, енцефаліт Бікерстафа, генералізований міоклонус і гострий поперечний мієліт; енцефаліти, включно з лімбічним енцефалітом і змішаним енцефалітом з різними рентгенологічними даними; гострі цереброваскулярні захворювання, включно з ішемічними інсультами (від 1,3 до 14,7 % пацієнтів з COVID-19), геморагічними інсультами (0,5–1,3 %), церебральним венозним тромбозом. Порушення периферичної нервової системи, про які повідомлялося при COVID-19, були такими: синдром Гієна — Барре і його варіанти, включно із синдромом Міллера — Фішера, черепним поліневритом і лицьовою диплегією, ізольована окорухова невропатія та міопатія критичного стану. Розтин пацієнтів із COVID-19, які померли від інсультів, показав наявність вірусу в мозку. У пацієнтів з COVID-19 з енцефалітом була позитивна полімеразна ланцюгова реакція на вірус у спинномозковій рідині [22].
Ураження печінки при COVID-19
COVID-19 також викликає ураження печінки. За допомогою конфокальної мікроскопії високої роздільної здатності в недавньому дослідженні були виявлені віруси білків SARS-CoV-2 у синусоїдальних ендотеліальних клітинах печінки (LSEC) з тканин пацієнтів із COVID-19. Крім того, рецептори лектину С-типу L-SIGN з високою експресією на LSEC і запальних ендотеліальних клітинах були ідентифіковані як рецептори інфекції SARS-CoV-2. Після вірусної інфекції в пацієнтів з початком перебігу COVID-19 виникає цитокіновий шторм, особливо підвищена секреція прозапальних цитокінів IL-1b і IL-6. Нещодавні дослідження показали, що дисфункція LSEC поширена серед хворих із COVID-19. Також повідомлялося, що передача сигналів через IL-6 і IL-1b рецептори в LSEC призводить до ендотеліопатії, формування дисфункції ендотелію та ураження печінки в пацієнтів з COVID-19. У пацієнтів з постковідним синдромом реєстрували підвищені рівні маркерів коагулопатії, дисфункції ендотелію, оксидативного стресу й ураження печінки (АЛТ, АСТ, білірубін, кисла й лужна фосфатаза, нітротирозин). Кореляційний аналіз показує, що рівень IL-6 позитивно корелює з рівнем маркерів ендотеліальної дисфункції (білок — рецептор протеїнкінази С (EPCR), eNOS, гомоцистеїн, D-димер). Активація транссигнального шляху з рецептора IL-6 на LSEC призводить до коагулопатії, підвищення експресії EPCR та iNOS, активації адгезії тромбоцитів до LSEC, а також активації позаклітинних механізмів апоптозу. Ці порушення блокуються розчинним глікопротеїном 130 (sgp130), селективними інгібіторами JAK-кіназ, відновленим глутатіоном [4, 7, 10, 22].
Ураження нирок при COVID-19
За даними звіту за 2020 р., у ЄС хронічна ниркова недостатність (ХНН) входить до числа основних супутніх патологій у померлих від COVID-19, становлячи 23,1 % і перебуваючи за частотою на 4-му місці після артеріальної гіпертензії (66 %), цукрового діабету 2-го типу (29 %) та ішемічної хвороби серця (ІХС) (27,9 %). При метааналізі 1389 спостережень встановлено значущий зв’язок між хронічною хворобою нирок (ХХН) і тяжкістю перебігу COVID-19 із відношенням шансів 3,03 (95% ДІ 1,09–8,47). Іншими дослідженнями (1203 пацієнти) показано, що COVID-19 викликає гостру ниркову недостатність, що включає дисфункцію ендотелію, запалення, некроптоз, закупорку капілярів, порушення фільтраційної та секреторної функції. Однак механізм пошкодження нирок при COVID-19 поки не зрозумілий. До цього часу встановлено, що для входження в клітину-мішень SARS-CoV-2 використовує АПФ-2. Ця сполука являє собою карбоксипептидазу, експресовану на клітинах нирки, яка розщеплює антіотензин-1 (АТ1) на АТ 1–9 і антіотензин-2 (АТ2) — на АТ 1–7. Тим самим АПФ-2 протидіє вазоконстрикторним, проліферативним і фіброзуючим ефектам АТ2, генерованим АПФ. SARS-CoV-2 інфікує насамперед і переважно клітини респіраторного тракту, оскільки АПФ-2 експресується на альвеолярних епітеліальних клітинах I і II типу в нижніх відділах легень. SARS-CoV-2 може зв’язуватися з АПФ-2 і через спайковий глікопротеїн, що експресується на вірусній оболонці. Після проникнення в альвеолярні клітини SARS-CoV-2 використовує ендогенний транскрипційний механізм для реплікації й поширення по всій легені. Однак АПФ-2 широко представлений також і на клітинах інших органів, зокрема серця, печінки, шлунково-кишкового тракту й нирок, що дає можливість вірусу пошкоджувати і ці органи [3, 15, 20, 21].
Стратифікація органів людини за рівнем високої та низької експресії АПФ-2 дозволяє говорити про дуже високу вразливість нирки до інфекції SARS-CoV-2. При цьому найбільшою мірою (~82 %) АПФ-2 експресується на епітелії проксимальних канальців, менше — на вставних клітинах збірних трубочок, епітелії дистальних канальців, гломерулярних парієтальних клітинах і подоцитах. Припускається, що вірус може проникати в нирку, зв’язуючись спочатку з АПФ-2 на подоцитах, а потім поширюючись у канальцеву рідину і далі в клітини проксимальних канальців. Мультиорганний і, зокрема, нирковий тропізм SARS-CoV-2 розглядається багатьма авторами як фактор, що лежить в основі пошкодження нирок при COVID-19. При дослідженні біоптатів нирок пацієнтів, які померли від COVID-19, у 60 % випадків виявили присутність у них РНК SARS-CoV-2. При гострому ушкодженні нирок РНК вірусу визначалася частіше, ніж у випадках, що перебігали без гострого ушкодження нирок.
Отримані дані дозволяють зробити висновок про кореляцію між екстрареспіраторним, і зокрема нирковим, тропізмом SARS-CoV-2 і тяжкістю перебігу COVID-19. Вважають, що ураження нирок при COVID-19 обумовлене нефротропним ефектом вірусу і його цитопатичним впливом на канальцевий епітелій паралельно з легеневим. Дисфункція нирок, що виникає при цьому, може посилювати запальний процес у легенях у рамках поєднаного ураження. З одного боку, виділення циркулюючих запальних факторів (наприклад, TNF-α та інтерлейкінів) при запальній реакції в легенях може вести до додаткового пошкодження нирки, а з іншого — пошкодження ниркового епітелію може посилювати легеневий процес і призводити до пошкодження інших органів. У якийсь момент такий взаємозв’язок викликає необоротний цитокіновий шторм, що самопосилюється, який швидко індукує поліорганну недостатність і смерть. Крім того, локальне запалення у відповідь на ушкодження й загибель ниркових клітин посилює гостре ушкодження нирок та ініціює ушкодження інших органів. Додаткову роль у розвитку гострого ушкодження нирок відіграє і обумовлена тяжким ушкодженням легень тривала вентиляційна підтримка. Вона супроводжується ризиком розвитку сепсису з вираженою гіпотензією і потребою в інотропних препаратах. Персистуюча гіпотензія і вазоконстрикція, що виникають у кінцевому підсумку, можуть індукувати або посилювати гострий тубулонекроз як морфологічний субстрат гострого пошкодження нирок [15, 18].
Нарешті, існує прямий доказ цитопатичної дії SARS-CoV-2 на проксимальний канальцевий епітелій. У більшості хворих з тяжким і вкрай тяжким перебігом COVID-19 виявлено ознаки проксимальної канальцевої дисфункції, якій на структурному рівні відповідали гострий тубулонекроз із втратою щіткової облямівки й значним зниженням у ній експресії мегаліну. При трансмісійній електронній мікроскопії в ендоплазматичному ретикулумі проксимальних канальців були ідентифіковані частки, які нагадують коронавірус, що може свідчити про пряму паренхіматозну інфекцію епітелію канальців і подоцитів. У пацієнтів у постковідному періоді виявлено значне підвищення молекулярних маркерів ураження нирок — цистатину С (підвищений при тубулярній дисфункції, є предиктором поганого результату в гетерогенній групі пацієнтів без початкової олігурії), молекули ушкодження нирок 1 (KIM-1) (трансмембранний глікопротеїн з ектодоменом, що містить Ig-подібний і муциновий домен, синтезується в дуже високій концентрації епітеліальними клітинами проксимальних канальців після ішемічного або токсичного пошкодження).
Порушення енергетичного обміну і мітохондріальна дисфункція при COVID-19
Інфекція COVID-19 має перебіг з вираженим порушенням оксигенації крові, пошкодженням альвеол, мітохондрій і роз’єднанням окисного фосфорилювання (клінічні спостереження 797 пацієнтів за листопад і грудень 2020 року). Єдиним шляхом вироблення енергії для життєво важливих процесів при тяжкому перебігу захворювання є анаеробне окиснення глюкози, яке насилу компенсує потреби організму в енергії. Ціною такому шляху вироблення енергії є виражений ацидоз зі стрімким зростанням лактатдегідрогенази венозної крові, протонів водню в цитоплазмі або лактату в артеріальній крові, безпосередньо корелюючи з розвитком гострого респіраторного дистрес-синдрому. Результат — загибель клітини.
Також було виявлено підвищений рівень кетонових тіл, зокрема 3-гідроксибутирату, який відіграє роль посередника сигнальних шляхів апоптозу, запалення й оксидативного стресу [17, 19, 22]. Підвищення 3-гідроксибутирату у хворих на COVID-19 супроводжується зниженням рівня ліпопротеїнів високої щільності та α-токоферолу й підвищенням рівня тригліцеридів у плазмі крові. Також нами було встановлено дискоординацію в циклі Кребса, про що свідчило значне зниження рівня цитрату, малату й ізоцитрату в еритроцитах пацієнтів із COVID-19. Причому ці порушення зберігалися протягом місяця після гострого періоду захворювання. Також було встановлено депривацію компенсаторного шунта енергії — малат-аспартатного човникового механізму вироблення енергії в умовах гіпоксії. У крові пацієнтів було встановлено зниження рівня α-кетоглутарату, малату, глутамату на тлі зниження активності малатдегідрогенази.
Захворювання на COVID-19 супроводжується також порушенням транспорту жирних кислот у мітохондрії, про що свідчило зниження рівня L-карнітину й підвищення рівня вільних жирних кислот. Багато авторів вказують на формування вторинної мітохондріальної дисфункції. При розтині пацієнтів, які померли від COVID-19, у тканинах легень методом електронної мікроскопії було виявлено велику кількість мітохондрій з порушенням ультраструктури [9, 11, 18, 19].
Нині існує узагальнене поняття «мітохондріальна дисфункція». Це типовий патологічний процес, який не має етіологічної та нозологічної специфічності. Розвиток мітохондріальної дисфункції призводить до порушення зворотного захоплення медіаторів (катехоламінів, дофаміну, серотоніну), іонного транспорту, генерації і проведення імпульсу, синтезу білка de novo, процесів трансляції та транскрипції; також активізуються «паразитарні» енергопродукуючі реакції, що призводить до суттєвого зменшення енергетичних запасів нейрональної клітини. Крім того, під дією гідроксил-радикалу відбувається відкриття мітохондріальних пор з експресією та виходом у цитозоль проапоптотичних білків. Відкриття пор відбувається за рахунок окиснення тіольних груп цистеїнзалежної ділянки білка внутрішньої мембрани мітохондрій (аденозинтрифосфат (АТФ)/аденозиндифосфат (АДФ) антипортер), що перетворює його на проникний неспецифічний канал-пору. Відкриття пор перетворює мітохондрії з «електростанцій» на «топку» субстратів окиснення без утворення АТФ. Точними біохімічними дослідженнями було встановлено, що порушення кисневого режиму тканин, гіперпродукція ексайтотоксичних амінокислот, зниження «нормальної» акумуляції Са2+ мітохондріями, пошкодження мембрани мітохондрій АФК посилює відкриття пор і вивільнення апоптогенних білків з пошкоджених мітохондрій [23].
Особливості комплексної терапії при постковідному синдромі.
Метаболітотропна терапія
Коронавірусна хвороба асоціюється з вираженим запальним процесом, у тому числі цитокіновим штормом. Останнім часом вчені все більше уваги приділяють ролі автоімунних механізмів у патогенезі COVID-19, особливо щодо механізмів розвитку ускладнень даної патології, найнебезпечнішим з яких є гострий респіраторний дистрес-синдром, який розвивається в 15–33 % хворих. Вважається, що однією з головних ланок його патогенезу є каскад цитокінових реакцій (гіперцитокінемія IL-1β, IL-2, IL-6, IL-7, IL-8, IL-17, IFN-γ, G-CSF, MCP1, TNF-α тощо), який умовно називається цитокіновим штормом і виникає в організмі хворого внаслідок надмірної активності нейтрофілів і їх здатності утворювати позаклітинні нейтрофільні пастки (NETs) [23, 24–27].
З цього випливає логічне питання про те, яку роль у патогенезі COVID-19 відіграють ейкозаноїди, які є медіаторами запальної реакції та нерозривно пов’язані з сигнальними каскадами, що реалізуються завдяки цитокінам та іншим сигнальним молекулам. Передбачається, що ейкозаноїди, особливо простагландин Е2, виконують одну з провідних функцій розвитку автоімунних і запально-деструктивних процесів при COVID-19 [2, 10, 19, 22, 25]. Запалення при вірусній інфекції призводить до оксидативного стресу, вторинної мітохондріальної дисфункції, енергодефіциту й розвитку лактат-ацидозу в клітині. Так, запалення при COVID-19 супроводжується розвитком оксидативного стресу в органах і тканинах. При цьому відбувається утворення великої кількості активних форм кисню й монооксиду азоту (NO), вільних радикалів і продуктів пероксидації ліпідів і білків. Надлишок АФК і NO в умовах антиоксидантної недостатності призводить до окисної модифікації ліпідів, нуклеїнових кислот і білків. Окисна модифікація білкових фрагментів рецепторів, іонних каналів, синаптичних структур нейрона призводить до порушення генерації, утворення, провідності нервового імпульсу, синаптичної передачі і, як наслідок, до погіршення функції клітин [23].
/16_m.jpg)
Відомо також, що під впливом АФК у клітині відбувається активація експресії редокс-чутливих генів, одні з яких необхідні для захисту клітин від токсичних ефектів окиснювального стресу, а інші при надлишку АФК ініціюють апоптоз. У розвиток багатьох захворювань великий внесок робить також така патогенетична ланка, як ішемія. Її прямим наслідком є порушення кисневого режиму тканин, різке зниження аеробної продукції АТФ і його дефіцит, активація анаеробного гліколізу й формування метаболічного лактат-ацидозу, зміщення рН у кислу сторону, що призводить до зниження активності ферментів і активації багатьох патохімічних реакцій. Енергодефіцит гальмує роботу синапсів, іонних каналів, підвищується пасивна проникність мембран для Са++. Надалі формується вторинна мітохондріальна дисфункція, і мітохондрії з «електростанцій» клітини, що виробляють АТФ, стають джерелами АФК і проапоптичних білків [23]. У крові пацієнтів із COVID-19 на різних стадіях захворювання виявляється збільшення маркерів оксидативного стресу — нітротирозину й карбонілованих білків. Паралельно реєструють порушення в тіол-дисульфідній системі — підвищення рівня окиснених тіолів, окисненого глутатіону, пригнічення активності глутатіон-залежних ферментів на тлі дефіциту відновленого глутатіону. Усе це призводить до пошкодження мембран клітин і клітинних органел АФК вільними радикалами й продуктами пероксидації, що, у свою чергу, веде до порушення функції і загибелі клітин на кшталт апоптозу або навіть некрозу.
/16_m2.jpg)
Останнім часом з’явилися дані щодо ініціювання піроптозу при COVID-19 [19, 21, 22]. Піроптоз — це запрограмований прозапальний вид загибелі лейкоцитів, насамперед моноцитів і макрофагів, що індукується протеазою каспазою-1 або іншими каспазами. Відомо, що ініціація піроптозу відбувається при попаданні в цитоплазму агентів бактеріального або вірусного походження, наприклад флагеліну (бактеріальний білок, що самоорганізується в порожнисті циліндричні структури й здатний зв’язуватися з рецептором TLR5). TLRs — це білкові рецептори до антигенів, що включають понад 20 підродин (NOD1, NOD2, NLRP1, NLRP2, NLRP3, NLRС4 тощо) [10]. При зв’язуванні з ними ініціюється вироблення прозапальних цитокінів: TNF, INF-α/β, IL-6, IL-8, IL-12, і насамперед IL-1β, IL-18. NLRP1, NLRP2, NLRP3, NLRС4 беруть участь у формуванні білкових комплексів — інфламасом, які, у свою чергу, активують каспазу-1, що збирається в активну форму з двох гетеродимерів. Під впливом каспази-1 відбувається утворення пор (шляхом розщеплення інгібітору білка GSDMD (Gasdermin D), фрагмент якого GSDMD-N вбудовується в мембрану з формуванням перфорацій у плазматичній мембрані діаметром до 10–14 нм, що призводить до осмотичного набухання клітини й лізису). Також відбувається фрагментація ДНК, яка активувалася ендонуклеазами. Цитокіни, які вивільняються з клітини, що гине, активують макрофаги, Т-лімфоцити, NK-клітини. Отже, клітина, що гине, стає фактором мобілізації та атракції нових лейкоцитів, необхідних для ефективної боротьби з інфекційним агентом [18].
Також встановлено, що піроптоз супроводжується зміною експресії білків теплового шоку (HSP). Активація оксидативного стресу при COVID-19, а також дефіцит відновлених інтермедіатів тіол-дисульфідної системи призводять до формування ендотеліальної дисфункції. Це підтверджується зниженням експресії ендотеліальної синтази монооксиду азоту й активності eNOS у крові пацієнтів. Усе це теоретично обґрунтовує перспективність застосування в комплексній терапії COVID-19 засобів метаболітотропної терапії.
Статини
Статини широко застосовуються як ефективні засоби профілактики й лікування пацієнтів з гіперхолестеринемією та ішемічною хворобою серця завдяки їх гіполіпідемічній і плейотропній протизапальній, антиоксидантній, антитромботичній та імуномодулюючій дії. Статини сьогодні розглядаються як засоби додаткової терапії для корекції ендотеліальної дисфункції при COVID-19. Однак використання статинів також може індукувати експресію АПФ-2, що може збільшити проникнення вірусу в клітину. Метааналіз (523 пацієнти) виявив позитивний ефект статинів у пацієнтів з COVID-19 (зниження лабораторних маркерів дисфункції ендотелію та ураження міокарда). Це відповідає даним нещодавнього звіту про підвищення ефективності терапії статинами в зниженні ризику летальності при COVID-19. У дослідах на культурі ендотеліальних клітин людини встановлено, що запалення й оксидативний стрес, індуковані COVID-19, пригнічуються аторвастатином, який підвищує експресію KLF2. Цей факт обґрунтовує перспективність застосування активаторів KLF2 для корекції ендотеліальної дисфункції, пов’язаної з COVID-19. Також варто відзначити, що багато авторів скептично ставляться до терапевтичної цінності статинів при постковідному синдромі [2, 3, 12].
Метформін
Загальновідомо, що в пацієнтів із цукровим діабетом 2-го типу (ЦД2) COVID-19 частіше перебігає в тяжкій і критичній формі, і вони частіше гинуть від ускладнень з боку серцево-судинної системи (інсульти, інфаркти міокарда) порівняно з пацієнтами, які не мають в анамнезі ЦД2. Гіперглікемія та активація поліолового метаболізму глюкози ще більше посилює наявне формування ендотеліальної дисфункції в пацієнтів із COVID-19. Плейотропні ефекти метформіну контролюють гіперглікемію, інгібують проникнення вірусу й зменшують запалення після інфекції SARS-CoV-2. Поліфармакологічний профіль метформіну робить його перспективним лікарським засобом-кандидатом для перепрофілювання для зниження цитокінового шторму в пацієнтів з діабетом і COVID-19. Отже, метформін може бути включений у комплексну терапію COVID-19 у пацієнтів із ЦД2 [2, 3].
Інгібітори SGLT2
Інгібітори SGLT2 розглядаються як перспективні лікарські препарати для лікування серцево-судинних захворювань у силу їх кардіо-, ендотеліопротективних ефектів та антиапоптичної дії. Інгібітори SGLT2 знижують летальність в експериментальних тварин з інфарктом міокарда, хронічною серцевою недостатністю зі зниженою фракцією викиду або зі збереженою фракцією викиду. У пацієнтів із COVID-19 із частими ускладненнями спостерігаються серцева недостатність і розвиток серцево-судинних захворювань, що обґрунтовує подальше вивчення інгібіторів SGLT2. Також експериментально доведено ендотеліопротективну активність інгібіторів SGLT, що обґрунтовує проведення клінічних випробувань при COVID-19 у пацієнтів із ЦД2 [1, 7, 11, 15, 18].
Модулятори ренін-ангіотензинової системи. Інгібітори АПФ-2, блокатори АТ2
Відома роль АПФ-2 у проникненні вірусів SARS-CoV і SARS-CoV-2 у клітину. AПФ-2 експресується в багатьох основних органах/тканинах, включно із серцем, легенями, нирками, при COVID-19. Розчинна форма АПФ-2 циркулює в крові пацієнтів із COVID-19 і може бути інформативним лабораторним маркером захворювання. SARS-CoV-2 викликає порушення балансу AПФ/AПФ-2 та активацію ренін-ангіотензин-альдостеронової системи, що в результаті призводить до прогресування COVID-19, особливо в пацієнтів із супутніми причинами, такими як гіпертонія, цукровий діабет і серцево-судинні захворювання, і запускає механізми пошкодження органів-мішеней. Експресія AПФ-2 може сприяти патогенності SARS-CoV-2. Спочатку передбачалося, що інгібітори АПФ або блокатори рецепторів АТ2 — широко застосовувані групи антигіпертензивних препаратів — можуть підвищити вразливість до SARS-CoV-2 за рахунок підвищення експресії AПФ-2. Подальші дослідження не підтвердили цю гіпотезу, було встановлено, що їх відміна призводить до серйозних ускладнень з боку серцево-судинної системи. У дослідженнях на 659 пацієнтах із COVID-19 було встановлено, що до 30-го дня дослідження вижили й виписалися з лікарні 91,8 % пацієнтів, які припинили прийом інгібіторів АПФ або блокатори АТ2 рецепторів, і 95 % — які приймали ці препарати. У групі пацієнтів, які приймали блокатори рецепторів АТ2, вірогідно знижувалися в крові маркери ендотеліальної дисфункції. Іншими дослідженнями виявлено, що прийом АТ2-блокаторів знижує опосередковану VEGF-A проангіогенну сигнальну гіперпроникність ендотелію при COVID-19. Захисна роль агонізму ACE2 у пригніченні ангіогенезу й підтримці цілісності ендотелію при COVID-19 потребує ретельного вивчення [14].
Тоцилізумаб
Активований цитокіновий шторм і передача сигналів IL-6 спостерігалися при ендотеліальній дисфункції під час бактеріальної інфекції та інфекції SARS-CoV-2. IL-6 починає впливати на ендотеліальні клітини, збільшуючи проникність судин, адгезію формених елементів крові і активуючи каскад коагуляції. Рівень IL-6 у пацієнтів з COVID-19 був настільки ж підвищений, як і при інших синдромах вивільнення цитокінів (сепсис або гострий легеневий дистрес-синдром). Лікування тоцилізумабом, специфічним блокатором рецептора IL-6, супроводжувалося зниженням маркерів запалення і дисфункції ендотелію в пацієнтів із COVID-19. Також встановлено, що призначення тоцилізумабу знижує ризики ішемічного ушкодження міокарда при COVID-19 (зниження в крові пацієнтів МІ-КФК і ST2 білка). Виявлено, що тоцилізумаб пригнічує експресію маркерів старіння (p21 і p16), утворення АФК, а також адгезію лейкоцитів, опосередковану ендотеліальними молекулами адгезії. Тоцилізумаб також пригнічує формування ендотеліальної дисфункції, збільшуючи товщину глікокаліксу, знижуючи маркери оксидативного стресу й апоптозу та підвищуючи експресію eNOS [2, 11, 14, 15].
Анакінра (РАІЛ)
IL-1β є важливим цитокіном, що вивільняється під час цитокінового шторму при COVID-19, а також при постковідному синдромі. Анакінра, що блокує рецептори IL-1β, виявляє кардіопротективну й ендотеліопротективну дію. Також є дані про нейропротективну дію РАІЛ. Анакінра та РАІЛ збільшують виживання інфікованих мишей. Анакінра добре переноситься пацієнтами з COVID-19 і дає обнадійливі результати, особливо в пацієнтів з концентрацією С-реактивного білка > 100 мг/л. РАІЛ нормалізує глутатіонову ланку тіол-дисульфідної системи головного мозку, підвищує експресію HSP70, гальмує нейроапоптоз [14].
Інгібітори JAK
Шлях JAK/STAT є канонічним шляхом виникнення запалення. Запальні цитокіни, такі як IL-6 і IL-1β, індукують фосфорилювання JAK і STAT. Після цього STAT транслокуються в ядро клітини, прискорюючи експресію запальних цитокінів, посилюючи повторну хвилю цитокінового шторму. Інгібітори JAK/STAT, такі як руксолітиніб, тофацитиніб і барицитиніб, можуть інгібувати сигнальний каскад цитокінів. У даний час були проведені клінічні випробування з метою оцінки безпеки й ефективності інгібіторів JAK/STAT [1, 14, 19].
Сенолітики
Вірус-індуковане старіння є патогенним тригером ендотеліальної дисфункції. Рівні маркерів старіння, такі як PAI-1, p21 і сиртуїн-1, в ендотеліальних клітинах значно підвищені. Сенолітичні препарати, такі як навітоклакс і комбінація кверцетину/дазатинібу, вибірково знищують клітини, що старіють, і зменшують запалення в тварин, інфікованих SARS-CoV-2. Терапевтичний потенціал сенолітиків у пацієнтів з COVID-19 потребує подальшого вивчення [14].
L-аргінін
COVID-19 ініціює формування ендотеліальної дисфункції, у якій важливу роль відіграє порушення у системі «L-аргінін — eNOS — NO». Препарати L-аргініну впливають на ендотеліальну дисфункцію, оскільки є субстратом утворення NO в ендотеліальних клітинах. Також встановлено, що L-аргінін має антиоксидатну й протизапальну дію, знижує рівень атерогенних ліпопротеїдів. У рандомізованому клінічному дослідженні додаткова терапія L-аргініном значно знижує тривалість госпіталізації в пацієнтів з постковідним синдромом, поліпшує показники ЕКГ [15, 20].
Флувоксамін
Нещодавні рандомізовані клінічні випробування показали, що лікування флувоксаміном (селективний інгібітор зворотного захоплення серотоніну) зменшує потребу в тривалій госпіталізації пацієнтів з COVID-19, знижує летальність і дає обнадійливий результат у перші 15 днів після захворювання. Флувоксамін також здатний збуджувати рецептори сигма-1 (S1R), які підвищують експресію IL-6 і одночасно значно посилюють експресію eNOS. Також показано, що стимуляція S1R-1 гальмує апоптоз ендотеліальних клітин, знижує ризики тромбоутворення й серцево-судинних катастроф. Ці дані свідчать про те, що у флувоксаміну можуть бути розширені показання до застосування, зокрема у складі комплексної терапії COVID-19. Усе це вимагає додаткових досліджень [5, 9, 14, 18, 20].
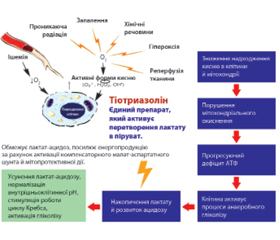
Тіотриазолін
Тіотриазолін має імуномодулюючу, протизапальну, антиоксидантну, протиішемічну, кардіопротекторну й гепатопротекторну активність [24–27]. Ефективність Тіотриазоліну за цими видами активності доведена як на доклінічному, так і на клінічному етапах дослідження й підтверджена більш ніж 20-річною історією застосування в охороні здоров’я країн пострадянського простору. Основний фармакологічний ефект Тіотриазоліну — антиоксидантний. У літературі накопичено численні дані щодо механізмів оксидативного стресу і його ролі в нормальному й патологічному функціонуванні клітин, проте, крім головного субстрату переокиснення — молекул біомембран і ядерного хроматину, АФК викликають окиснювальну модифікацію білків.
Вважається, що в стані окиснювального стресу атаці АФК піддаються не ліпіди, а насамперед білки плазматичних мембран. Підтвердженням цього може бути феномен, названий Бергельсоном молекулярною пам’яттю [23, 27]. Суть його в тому, що багато короткострокових подій, які перебігають у білковій молекулі клітинної мембрани, впливають на довготривалі параметри функціонування мембранного бішару. При впливі відповідного агента на мембранний білок-рецептор конформація останнього змінюється й надалі індукує зміни білка — ліпідних контактів, стану ліпідів, що оточують білок. Ці зміни стану ліпідів зберігаються і після відщеплення ліганду від рецептора, тобто служать способом закріплення рецептора в збудженій конформації. Отже, «пам’ять» ліпідів забезпечує посилення сигналу, що передається із зовнішнього середовища на клітинну мембрану.
/17_m.jpg)
Підтвердженням первинності перекисного окиснення білка є наявність виражених змін при окиснювальному стресі в ділянці анулярних ліпідів, що свідчить про провідну роль окисної модифікації білків у деструкції клітинних білкових структур мембрани — іонних каналів, рецепторів тощо. Тіол-дисульфідна система реагує на будь-який вплив внутрішнього або зовнішнього характеру зміною свого окисно-відновного стану [28]. Цей стан можна охарактеризувати співвідношенням концентрації -SH- і -SS-груп (SH/SS) — тіосульфідне співвідношення (ТДС). Зміна редокс-рівноваги в цій системі має різноспрямований (фазовий) характер і залежить від сили й тривалості діючого фактора. Іншими словами, початкова зміна в ТДС, що характеризується зсувом редокс-рівноваги у бік відновлення, замінюється зміною протилежної спрямованості — зсувом редокс-рівноваги в бік окиснення [23, 26, 29]. Це можна розглядати як ознаку окиснювального ушкодження білкової молекули. Зсув ТДС у бік окиснених еквівалентів призводить до порушення фізіологічного механізму розвитку пострецепторного сигналітету.
Одним з механізмів формування десенситизації рецепторів є окиснювальна модифікація білкових структур рецептора, утворення карбонільних груп під дією надлишку АФК. Крім того, втрата чутливості рецепторів відбувається за рахунок непрямої взаємодії цитотоксичних форм NO (S-, N-, O-нітрозування) з тіольними, фенольними, гідроксильними й аміногрупами білків. Нітрозування функціональних груп ДНК призводить до пригнічування експресії білків, які є рецепторами. Подібні механізми є основою формування толерантності (мітридатизму) до лікарських препаратів. Вищевикладене обґрунтовує включення антиоксидантів до комплексної терапії багатьох захворювань для переривання АФК-/NO-залежних механізмів формування медикаментозної резистентності.
/17_m2.jpg)
Найбільш вивченим із цієї точки зору є тіотриазолін [23, 25, 27]. Антиоксидантна дія тіотриазоліну полягає в тому, що препарат активує антиоксидантні ферменти — супероксиддисмутазу, каталазу, глутатіон-пероксидазу, сприяє більш економному витрачанню ендогенного антиоксиданту — α-токоферолу, нормалізує тіол-дисульфідну систему й підвищує рівень відновленого глутатіону, гальмує окиснювальну модифікацію білків, ліпідів, нуклеїнових кислот, знижуючи утворення маркерних продуктів цих процесів — нітротирозину, 8-гідроксигуаніну, карбонілованих білків і малонового діальдегіду.
За силою антиоксидантної дії тіотриазолін вірогідно перевершує в кілька разів такий природний антиоксидант, як α-токоферол, а також відомі синтетичні антиоксиданти — дибунол (іонол), емоксипін, мексидол, ацетилцистеїн. Дослідженнями in vitro було показано, що тіо–триазолін у діапазоні концентрацій 10–5–10–7 М знижує концентрацію таких АФК, як супероксидрадикал (О2–) і пероксинітрит (ОNОО–). Подібну дію тіотриазолін виявляє завдяки тому, що в його структурі міститься тіольна група, яка наділяє всю молекулу високими відновними властивостями й здатна приймати електрони від АФК. У результаті сірка в тіольній групі переходить із двох- до чотиривалентного стану. Тіотриазолін зменшує утворення АФК у біоенергетичних реакціях у мітохондріях, а також у ксантиноксидазній реакції. Найбільш вивчено протективну дію тіотриазоліну щодо сульфгідрильних груп цистеїнових і метіонінових фрагментів білкових молекул. Тіотриазолін конкурує з цими структурами за супероксидрадикал, унаслідок чого запобігає як оборотній, так і необоротній їх модифікації [29].
Більш значуща за ефективністю дія тіотриазоліну реалізується щодо необоротної модифікації сульфгідрильних груп низки білкових молекул під дією АФК. Так, тіотриазолін гальмує утворення в білках необоротних сульфоксидів і сульфонових груп, які надалі легко піддаються окисненню. Здійснюючи гальмівну дію на необоротну окиснювальну модифікацію сульфгідрильних груп цистеїнових фрагментів білкових молекул, тіотриазолін нормалізує зрушення red-oxi-регуляції в умовах оксидативного стресу [24–28]. Насамперед тіотриазолін запобігає розвитку порушення рівноваги тіосульфідної системи при гіперпродукції АФК, забезпечуючи такі функції, як передача клітинного сигналу через рецепторно-іоноформний комплекс, зберігаючи активність білків, ферментів, факторів транскрипції і цілісність клітинних мембран. Тіотриазолін перешкоджає необоротній інактивації фактора транскрипції NF-κВ, захищаючи від надлишку АФК чутливі залишки цистеїну — Cys 252, Cys 154 і Cys 61 у його ДНК-зв’язуючих доменах.
Крім того, тіотриазолін може брати участь у відновленні цих груп при оборотній інактивації, беручи на себе роль Redox Faktor-1 [23]. Гальмуючи окисну інактивацію фактора транскрипції NF-κВ при надлишку АФК, тіотриазолін, можливо, посилює активацію експресії редокс-чутливих генів, відповідальних за синтез рецепторів білків.
Отже, у механізмі антиоксидантної дії тіотриазоліну можна виділити наступне: зменшуючи концентрацію АФК (супероксидрадикал і пероксинітрит) як за рахунок прямої взаємодії з ними, так і за рахунок гальмування шляхів їх утворення, даний препарат знижує ступінь окисної модифікації білкових структур рецепторів. Дослідженнями член-кор. І.С. Чекмана і проф. І.Ф. Бєленічева було встановлено, що тіотриазолін підвищує ефективність бета-адреноблокаторів, блокаторів АТ2 при артеріальній гіпертензії та ІХС за рахунок збереження чутливості адренорецепторів і ангіотензинових рецепторів в умовах активації оксидативного стресу при цих патологіях серцево-судинної системи [29, 30]. Тіотриазолін сприяє підвищенню рівня відновленого глутатіону, який регулює Red-/Oxi-механізми експресії генів, відповідальних за синтез ферментів, у тому числі тих, що регулюють прозапальні каскади — ліпоксигеназний і циклооксигеназний. Тіотриазолін може й безпосередньо брати участь у регуляції транскрипційної активності, запобігає розвитку порушення рівноваги тіосульфідної системи при гіперпродукції АФК, забезпечуючи такі функції, як передача клітинного сигналу через рецепторно-іоноформний комплекс, зберігаючи активність білків, ферментів, факторів транскрипції і цілісність клітинних мембран. Є дані, що тіотриазолін виявляє імуномодулюючу активність, підвищуючи рівень інтерферону, а також кількість Т-лімфоцитів [31].
У численних дослідженнях встановлено, що тіотриазолін виявляє протизапальну активність, перешкоджаючи необоротній інактивації фактора транскрипції NF-κB, і гальмує експресію прозапальних цитокінів IL-1β, IL-6, TNF-α, а також С-реактивного білка, iNOS [14, 18, 31, 32]. Тіотриазолін стабілізує мембрани базофілів, тучних клітин та еозинофілів, збільшує фагоцитарну активність макрофагів. З огляду на вищевикладені дані, що переконливо доводять негативну роль АФК, цитотоксичних інтермедіатів оксиду азоту та оксидативного стресу в механізмах запалення, болю й набряків, включення тіотриазоліну в комплекс лікування дає прогнозоване потенціювання дії засобів базової терапії. Крім цього, якщо враховувати низку серйозних побічних ефектів базових нестероїдних протизапальних препаратів (НПЗП) і аналгетичних ненаркотичних засобів, спрямованих на порушення тонких ланок метаболізму кардіоміоцитів, ендотеліоцитів, гепатоцитів тощо, уведення в комплексну терапію запальних захворювань антиоксидантів, у тому числі тіотриазоліну, може підвищити безпеку пропонованого медикаментозного лікування.
До дуже цікавих ефектів тіотриазоліну належить його захисна дія щодо ендотелію судин, що має велике значення при COVID-19, оскільки при даній патології неминуче розвивається ендотеліальна дисфункція. У звітах з доклінічного вивчення тіотриазоліну й дисертаційних дослідженнях показано, що тіотриазолін збільшує біодоступність NO, підвищуючи рівень SH-сполук, а також самостійно утворюючи нітрозотіольні комплекси з NO. Усе це захищає NO від взаємодій з активними формами кисню і його перетворення на цитотоксичний і прозапальний пероксинітрит. Тіотриазолін підвищує щільність ендотеліоцитів і проліферуючих ендотеліоцитів, збільшує експресію васкулоендотеліального фактора (VEFF) та ендотеліальної синтази монооксиду азоту [23, 27, 32].
Відомо, що COVID-19 призводить до ускладнень і порушує систему згортання крові й тромбоутворення. Тіотриазолін виявляє фібринолітичні й антиагрегантні властивості. Отримано численні дані, що при ішемії міокарда тіотриазолін у тромбоцитах значно підвищує активність глутатіонпероксидази, знижує накопичення продуктів окисної модифікації ліпідів, що, імовірно, призводить до зменшення в крові рівня тромбоксанів, які беруть участь у тромбоутворенні. Не виключається вплив тіотриазоліну на АФК-залежні механізми експресії тканинного плазміногену [23].
З огляду на ускладнення з боку серцево-судинної системи, спричинені впливом як самого коронавірусу, так і препаратів, що застосовуються при лікуванні COVID-19, актуальними є дані про кардіопротекторну дію тіотриазоліну, які були отримані в низці доклінічних і клінічних досліджень, а також унаслідок 20-річного досвіду застосування тіотриазоліну в кардіології. Тіотриазолін поліпшує показники ЕКГ, зменшує зону некрозу при експериментальному інфаркті міокарда, знижує летальність. Тіотриазолін посилює синтез АТФ, нормалізує дихальний ланцюг мітохондрій і підвищує утилізацію глюкози, вільних жирних кислот, глікогену в клітинах, обмежує малопродуктивний анаеробний гліколіз і запобігає розвитку лактат-ацидозу в кардіоміоцитах, нормалізує роботу ферментів циклу Кребса, а в умовах ішемії міокарда активує компенсаторний малат-аспартатний шунт енергії (продуктивніший і безпечніший, ніж анаеробний гліколіз) [23, 25, 26].
За силою кардіопротективної дії тіотриазолін перевершує такі відомі кардіопротектори, як мілдронат, L-карнітин, триметазидин (предуктал), рибоксин, цитофлавін, янтовіт, мітомін, коензим Q10, АТФ-Лонг. У клінічних дослідженнях за участю понад 1000 пацієнтів (у тому числі похилого віку) було показано позитивний вплив тіотриазоліну на стан кардіогемодинаміки при ІХС. Тіотриазолін помітно знижував загальний периферичний опір судин, вірогідно збільшував об’єм серцевого викиду з прогресивним зниженням витрат енергії міокардом. Поряд із цим у групі пацієнтів, які отримували тіотриазолін, підвищувалася толерантність до фізичного навантаження, що супроводжувалося помітним зростанням величини інотропного резерву міокарда [25, 27, 28].
Також тіотриазолін підвищував ефективність базової антигіпертензивної та антиангінальної терапії. На тлі призначення тіотриазоліну при гострому коронарному синдромі відбувалося вірогідне зниження смертності, пов’язане зі зменшенням кількості шлуночкових аритмій, більш швидке відновлення скорочувальної функції міокарда. Показано добру переносимість і безпеку курсового застосування (8 тижнів) тіотриазоліну в добовій дозі 600 мг для лікування ІХС, стабільної стенокардії напруги ІІ–ІІІ функціонального класу. Останні дані вказують також на нейротоксичний вплив SARS-CoV-2, який, зокрема, проявляється у вигляді гострого респіраторного дистрес-синдрому внаслідок токсичного пошкодження стовбура мозку, що призводить до розладу кардіореспіраторного центру й зупинки дихання [31–35].
Доклінічними дослідженнями було встановлено нейропротективну активність тіотриазоліну при гострому порушенні мозкового кровообігу. Так, введення тіотриазоліну приводило до зменшення летальності, підвищення щільності нейронів сенсомоторної зони кори головного мозку, гальмування нейроапоптозу, підвищення рівня аденозинтрифосфату й аденозиндифосфату в тканинах мозку й гальмування оксидативного стресу. Також введення тіотриазоліну приводило до зниження неврологічної симптоматики після моделювання гострих порушень мозкового кровообігу. Показано високу ефективність клінічного застосування тіотриазоліну при лікуванні судинної патології ока — транссудативних форм центральних хоріоретинальних дистрофій. Показано, що при включенні тіотриазоліну в комплексне лікування дітей з функціональною патологією центральної нервової системи поліпшення стану досягається в короткі терміни і дає віддалені добрі результати [23].
Тіотриазолін виявляє антиапоптичну активність, гальмуючи апоптоз кардіоміоцитів при хронічній серцевій недостатності, артеріальній гіпертензії, ІХС [23].
Медикаментозна терапія COVID-19 агресивна і має низку серйозних побічних реакцій з боку печінки й певні протипоказання (пацієнти з печінковою недостатністю, які перенесли гепатит, пацієнти похилого віку) [23, 25, 32]. Наприкінці 80-х років минулого століття було встановлено високу гепатопротекторну активність тіотриазоліну. Показано, що тіотриазолін приводить до нормалізації активності аланінамінотрансферази й аспартатамінотрансферази, лактатдегідрогенази, знижує тимолову пробу, підвищує рівень білка й знижує активність оксидативного стресу.
Показано, що застосування тіотриазоліну при лікуванні осіб з алкогольною хворобою печінки супроводжується позитивною динамікою клініко-біохімічної активності захворювання: регресією клінічних симптомів, значним зниженням вираженості цитолітичного синдрому, поліпшенням білково-синтетичної функції печінки. Включення тіотриазоліну з пірацетамом у комплексну терапію хворих на субкомпенсований цироз печінки приводить до істотного зниження симптомів печінкової енцефалопатії, поліпшуючи якість життя пацієнтів. Включення тіотриазоліну в лікування хворих на цироз печінки дає добрий терапевтичний ефект, зокрема призводить до нормалізації маркерів фібротичних процесів протягом 6 місяців [23].
Отже, тіотриазолін — вітчизняний препарат з імуномодулюючою, протизапальною, антиоксидантною, кардіопротекторною, гепатопротекторною дією, великим досвідом застосування в клінічній практиці та добре вивченим профілем безпеки — є перспективним засобом для профілактики інфікування й лікування (у складі комбінованої терапії) хворих на COVID-19. Усе це підтверджує доцільність клінічних випробувань тіотриазоліну для подальшого обґрунтування його застосування для профілактики інфікування коронавірусом і лікування коронавірусної хвороби, а також її ускладнень.
Нами проведені дослідження й отримані дані про ефективність застосування тіотриазоліну при його включенні до комплексної терапії постковідного синдрому.
Дослідження проводили на базі Університетської клініки ЗДМУ. У дослідженнях брали участь 15 відносно здорових добровольців і 62 хворі віком від 30 до 65 років з постковідним синдромом. З них 20 пацієнтів отримували базову терапію (антибіотики, антикоагулянти, ацетилсаліцилова кислота), а 42 хворі на тлі базової терапії додатково отримували тіотриазолін. 33 хворі отримували тіотриазолін у вигляді таблеток (по 200 мг), 9 пацієнтів — у вигляді супозиторіїв далмаксін по 0,2 г (діюча речовина — тіотриазолін) двічі на день протягом 30 діб. У дослідженнях було використано дві лікарських форми тіотриазоліну — таблетки й супозиторії. Використання супозиторіїв обумовлено тим, що пацієнти при лікуванні постковідного синдрому отримують значну кількість препаратів у таблетованій формі (у тому числі для лікування супутніх патологій), що може викликати незручності при дотриманні пацієнтами схеми прийому лікарських засобів. Крім того, для пацієнтів у тяжкому стані, тих, які знаходяться під кисневою маскою або ж на штучній вентиляції легень, такий шлях введення буде більш зручним. При цьому слід зазначити, що різниці в ефективності засобу тіотриазолін у різних лікарських формах — таблетки й супозиторії — немає (попередніми дослідженнями було доведено високу гепатопротективну ефективність тіотриазоліну саме у вигляді такої лікарської форми, як супозиторії). Критерієм включення в дослідження був позитивний ПЛР-тест на COVID-19; якщо ПЛР-тест був негативним — наявність IgM COVID-19 або IgG COVID-19 (при рентгенологічно підтвердженій пневмонії). Наявність пневмонії підтверджували за допомогою комп’ютерного або рентгенологічного дослідження органів грудної порожнини. Рівень ураження легень — до 45 % [36–38]. Пацієнти мали таку супутню патологію: цукровий діабет у стадії компенсації, артеріальну гіпертензію, ішемічну хворобу серця без серцевої недостатності. При надходженні та після закінчення лікування в усіх пацієнтів шприцом забиралась кров з ліктьової вени (10 мл), яку ділили для наступних видів дослідження — 1 мл уміщували у вакуумні пробірки з цитратом натрію для вивчення агрегації тромбоцитів, 0,5 мл — у вакуумні пробірки для аналізу лейкоцитарної формули крові. Решту використовували для отримання плазми методом центрифугування (20 хв при 1500 об/хв) на центрифузі Eppendorf centrifuge 5810 R (Німеччина) і додавання 0,129 моль/л цитрату натрію у співвідношенні до крові 1 : 9. Отриману плазму розливали по 0,5 мл по пробірках Eppendorf і зберігали в морозильній камері NZ-280/75A при –40 °С. У подальшому зразки розморожували й використовували для біо–хімічних досліджень — 0,2 мл, для імуноферментних досліджень — 0,1 мл.
Біохімічними методами визначали: С-реактивний білок — імунотурбодиметричним методом (набір виробництва Cormay, біохімічний аналізатор ACCENT-200, Польша); D-димер (набір виробництва «Вектор-Бест»), а також для встановлення гепатопротективної дії тіотриазоліну при постковідному синдромі проводилось визначення печінкових ензимів: АЛТ, АСТ, γ-глутамілтрансферази (ГГТ), рівня загального білірубіну (діагностичні набори виробництва Cormay, біохімічний аналізатор ACCENT-200, Польща); турбодинамічним методом проводилась ти–молова проба (НВП «Філісіт-Діагностика») [36 38].
Методом імуноферментного аналізу визначались наступні показники: IFN-γ (eBioscience (Bender MedSystems)); IL-4 (високочутливий, кат. номер BMS225HS, eBioscience (Bender MedSystems); IL-2 (високочутливий, кат. номер BMS221HS, eBioscience (Bender MedSystems); IL-17 (високочутливий (IL-17A hs), eBioscience (Bender MedSystems); eNOS (Cloud-Clone Corporation) — імуноферментним методом (імуноферментний аналізатор — Immunochem-2200, США).
Імунохемілюмінесцентним методом визначався феритин (набір виробництва Siemens, аналізатор — Immulate 1000, Велика Британія).
Коагулометричним методом визначалось міжнародне нормалізоване відношення (МНВ) (набір виробництва Diagon, Австрія, прилад — коагулометр CoagChrom 3003, Польща).
Паралельно проводили визначення агрегації тромбоцитів для оцінки гемостатичної функції тромбоцитів. Агрегаційна активність тромбоцитів досліджувалась шляхом турбідиметричного методу (оптична агрегометрія) за допомогою агрегометра Solar AP 2110 (Республіка Білорусь).
Дослідження рівня агрегаційної активності тромбоцитів — при внесенні індуктора агрегації АДФ (5,0 мкМ). Матеріал для дослідження: збагачена тромбоцитами цитратна плазма в об’ємі 1,0 мл. За два тижні до дослідження пацієнти припиняли прийом препаратів, що впливають на агрегацію тромбоцитів. Цільну кров відбирали в пластикову пробірку з 3,2% (0,109 М) або 3,8% (0,129 М) цитратом натрію у співвідношенні 9 : 1 або у вакуумні системи для взяття крові з 3,2% (0,109 М) цитратом натрію. Одразу ж після взяття крові вміст пробірки обережно перемішували перевертанням не менше за 5 разів без спінювання. Протягом 45 хв доставляли пробірку в лабораторію та центрифугували. Центрифугування зразка цільної крові проводили при кімнатній температурі (18–25 °С) протягом 5–7 хв при 1000 об/хв. Після завершення центрифугування відразу відбирали 1 мл збагаченої тромбоцитами плазми в чисту пластикову пробірку для подальшого дослідження. Отримання бідної тромбоцитами плазми (БТП) використовується як холоста проба (точка відліку). Для отримання БТП центрифугували зразок цільної крові при кімнатній температурі (18–25 °С) протягом 15 хв при 3000 об/хв. Після завершення центрифугування відбирали 1 мл БТП у чисту пластикову пробірку. Забір крові проводили тільки в вакуумні системи або пластикові пробірки з 3,8% цитратом натрію. Перед проведенням аналізу проводили попередній підрахунок клітин у плазмі на гематологічному аналізаторі або мікроскопічним методом, і відповідно до отриманих результатів збагачену тромбоцитами плазму розводили бідною тромбоцитами плазмою (від того ж пацієнта) так, щоб підсумкова кількість тромбоцитів в суміші становила 200–300 × 109/л. Як активатор агрегації використовували розчин АДФ з концентрацією 5,0 мкМ. Для приготування робочого розчину 4,7 мг АДФ додавали до 20 мл фізіологічного розчину, потім 1 мл отриманого розчину додавали до 9 мл фізіологічного розчину. Отримані результати вимірювали за відсотком світлопоглинання [36–38].
Дослідження показників рідкої крові (0,2 мл) проводили з використанням гематологічного автоматичного аналізатора Abacus 5 (Diatron, Угорщина). Досліджувались наступні показники: кількість лейкоцитів, лимфоцитів, моноцитів і тромбоцитів. При надходженні всі пацієнти пред’являли скарги на виражену слабкість, підвищену втому, серцебиття, підвищення температури тіла від 37,2 до 38,3 °С. Рівень ураження легень — до 45 %. Скарги на відсутність відчуття запаху й смаку мали 53,2 % пацієнтів, на кашель — 51,6 %, задишку — 45,1 %, діарею і біль у животі — у середньому 25,8 % . Пацієнти також відзначали коливання артеріального тиску, особливо ті, у яких є супутня артеріальна гіпертензія. Коливання відзначались, незважаючи на постійний прийом специфічної терапії (блокатори Са++-каналів, інгібітори АПФ, сартани, бета-адреноблокатори). Після лікування в групі пацієнтів, які приймали тіотриазолін, зникли скарги на серцебиття, артеріальний тиск стабілізувався (без додаткової корекції гіпотензивними препаратами), зникли слабкість і підвищена втома. Сатурація в 40 (95,2 %) пацієнтів основної групи підвищилась до 98–99 %. У контрольній групі в 9 (45 %) з 20 пацієнтів сатурація була на рівні 98 %.
Проведеними біохімічними й коагулометричними дослідженнями було встановлено, що у хворих з постковідним синдромом на тлі базисної терапії (антибіотики, антикоагулянти, ацетилсаліцилова кислота) порівняно з відносно здоровими пацієнтами спостерігалась підвищена концентрація С-реактивного білка й феритину на тлі зниженого МНВ і зниженої концентрації eNOS у плазмі крові. Однак у цій групі спостерігалось –вірогідне зни–ження С-реактивного білка порівняно з даними до початку лікування. При дослідженні вмісту D-димеру не було встановлено статистично вірогідних змін (референтні значення до 285 DDU). Введення в базисну терапію тіотриазоліну (протягом 1 місяця) приводило до нормалізації МНВ (вірогідне підвищення щодо показників пацієнтів при находженні на 97,2 % і щодо групи базисної терапії — на 50 %) і підвищення вмісту eNOS (вірогідне підвищення щодо показників пацієнтів при надходженні в середньому на 134,3 % і щодо групи базисної терапії — на 73,8 %), зниження D-димеру (вірогідне зниження щодо групи пацієнтів при надходженні на 32,3 % і щодо групи базисної терапії — на 16,1 %). Міжнародне нормалізоване відношення — це одне з досліджень на протромбін. За його допомогою визначається стан системи згортання крові в пацієнта. Даний білок є попередником тромбіну — білка, що стимулює формування тромбу. Зниження вмісту eNOS є ознакою дисфункції ендотелію. D-димер є найбільш специфічним маркером деградації фібринових згустків будь-якої локалізації, простіше кажучи, маркером інтенсивності й характеру процесів тромбоутворення. Збільшення концентрації D-димеру чітко й однозначно свідчить про активацію фібринолізу, чому, у свою чергу, обов’язково передує надмірне утворення нерозчинного фібрину, тобто тромбу. Введення в базисну терапію тіотриазоліну приводило до зниження феритину (вірогідне зниження щодо показників пацієнтів при находженні на 19,7 % і щодо групи базисної терапії — на 5,9 %). Показники С-реактивного білка статистично не відрізнялись від аналогічних значень контрольної групи хворих.
При визначенні агрегаційної активності тромбоцитів було встановлено, що у хворих з постковідним синдромом на тлі базисної терапії –порівняно зі здоровими пацієнтами спостерігалось підвищення агрегаційної активності тромбоцитів. Відсоток світлопоглинання становив у середньому 99,4 % проти 60 % у відносно здорових пацієнтів. Паралельно спостерігалось підвищення швидкості на 30-й секунді при збереженні нормальної кількості тромбоцитів — (380 ± 54,8) x 109/л. Показники відносно здорових пацієнтів не відрізнялись від референтних значень (агрегація тромбоцитів — 50–80 %; швидкість на 30-й секунді — 58–114 %; кількість тромбоцитів — 260–600 x 109/л). Введення в базисну терапію тіотриазоліну (протягом 1 місяця) приводило до зменшення агрегаційної активності тромбоцитів на 32 % і швидкості агрегації на 30-й секунді — на 54 %. Варто зазначити, що показники пацієнтів цієї групи знижувались щодо показників відносно здорових пацієнтів.
Нами отримано дані стосовно захисної дії тіотриазоліну щодо ендотелію судин, що має велике значення при COVID-19, тому що при даній патології неминуче розвивається ендотеліальна дисфункція. Відзначено, що формування ендотеліальної дисфункції при COVID-19 більш швидко відбувається в літніх пацієнтів, які приймають інгібітори АПФ. Ендотеліальна дисфункція є предиктором таких грізних захворювань, як інсульти й інфаркти міокарда. Загальновідомо, що NO є нестабільним, короткоживучим радикалом і для його стабілізації і подальшого транспортування передбачені такі механізми, як взаємодія з тіол–вмісними низькомолекулярними сполуками (глутатіон, цистеїн, метіонін) і відтворення стійких S-нітрозольних комплексів. В умовах дефіциту тіольних сполук при COVID-19 порушується транспорт NO, тому що він піддається атаці таких АФК, як супероксидрадикал і гідроксилрадикал з перетворенням у цитотоксичний продукт — пероксинітрит [23, 36]. При цьому спостерігається посилення формування дисфункції ендотелію. У звітах з доклінічного вивчення тіотриазоліну й у дисертаційних дослідженнях показано, що тіотриазолін підвищує біодоступність NO, підвищуючи рівень SH-сполук, а також самостійно утворюючи нітрозотіольні комплекси з NO. Усе це захищає NO від взаємодій з активними формами кисню і його перетворення в цитотоксичний і прозапальний пероксинітрит. Тіотриазолін підвищує щільність ендотеліоцитів, щільність проліферуючих ендотеліоцитів, підвищує експресію VEGF і eNOS. У клінічних дослідженнях показано, що комбінування тіотриазоліну й аргініну приводить до значного посилення ендотеліопротективної дії і дає протективний ефект відносно синтезу й транспорту NO, його біодоступності [37]. Також відомо, що тіотриазолін гальмує пошкодження ендотелію в зоні альтерації та індукує розвиток реакцій адаптації, зокрема перетворення плазміногену на плазмін з подальшим посиленням фібринолізу, що забезпечує лізис тромбів і нормалізацію кровообігу в зоні запалення. Тіотриазолін, знижуючи АФК-залежне пошкодження ендотелію в зоні альтерації, опосередковано зменшує трансформацію простагландину G2 у тромбоксан А2 під впливом тромбоксансинтетази тромбоцитів [23].
Також були отримані дані щодо антикоагулянтної дії тіотриазоліну. Відомо, що COVID-19 призводить до ускладнень і порушує згортання крові й тромбоутворення. Тіотриазолін виявляє антикоагулянтні й антиагрегантні властивості. Цей факт має підтвердження. Отримано численні дані, що при ішемії міокарда тіотриазолін у тромбоцитах значно підвищує активність глутатіонпероксидази, знижує накопичення продуктів окисної модифікації ліпідів, що, імовірно, приводить до зменшення в крові рівня тромбоксанів, які беруть участь у тромбоутворенні. Не виключається вплив тіотриазоліну на АФК-залежні механізми експресії тканинного плазміногену.
Одним з важливих компонентів постковідного синдрому є розвиток несприятливих побічних ефектів лікарських засобів, що застосовуються для лікування коронавірусної хвороби, а саме порушення гепатобіліарної системи та захисної функції печінки. У пацієнтів спостерігалось підвищення печінкових ферментів у плазмі крові, підвищення вмісту білірубіну. У 54 хворих з 57 при госпіталізації реєстрували астеновегетативний, холестатичний, больовий синдром в епігастрії і правому підребер’ї.
Введення в базисну терапію тіотриазоліну у вигляді таблеток (по 200 мг двічі на добу) або супозиторіїв (по 0,2 г двічі на добу) протягом 30 днів приводило до зменшення прояву цитолітичного синдрому, що проявлялось у зниженні АЛТ і АСТ на 56,4 і 67,1 % порівняно з показниками хворих при надходженні та на 48,5 і 61,0 % відповідно порівняно з групою хворих, які отримували базисну терапію. Важливо зазначити, що в групі хворих, які отримували базисну терапію, значення АСТ залишились підвищеними. Крім того, нормалізувалась білоксинтезуюча функція печінки, про що свідчила нормалізація показника тимолової проби на відміну від групи хворих, які отримували базисну терапію; також відбувалось зменшення загального білірубіну плазми крові й рівня ГГТ. У 40 хворих із 42 (95,2 %), які отримували курсом тіотриазолін, відзначалась позитивна динаміка, а саме зменшення астеновегетативного, холестатичного, больового синдрому в епігастрії та правому підребер’ї. У групі, що отримувала базову терапію, позитивна динаміка спостерігалась тільки в 9 хворих із 20 (45 %).
Згідно з результатами раніше проведених досліджень, механізм гепатопротективної дії тіотриазоліну полягає в його антиоксидантній, мембранопротективній і мітопротективній активності. Тіотриазолін здатний захищати ензими пентозофосфатного шунта, циклу трикарбонових кислот у гепатоцитах від їх окисного пошкодження, що забезпечує достатньо високий рівень енергетичних і пластичних процесів печінкової тканини [37]. Крім того, за рахунок наявності в молекулярній будові тіотриазоліну вільних SH-груп він здатний зв’язувати й інактивувати цитотоксичні деривати окисного стресу й метаболіти ксенобіотиків [36]. Тіотриазолін захищає мембрани мітохондрій печінки при токсичному гепатиті. Цей ефект підтверджується збереженням мембранного потенціалу мітохондрій і функціональним збереженням циклоспорин-А-залежної мітохондріальної пори. Мембраностабілізуюча активність тіотриазоліну реалізується за рахунок гальмування процесів перекисного окиснення ліпідів у мембранах ендоплазматичного ретикулуму печінки (зниження утворення МДА, гальмування біохемілюмінесценції) при токсичному ураженні печінки. Мембранопротективна активність тіотриазоліну проявляється також у здатності нормалізувати низку фізико-хімічних параметрів структури мембран (флуоресценція 1,8-аніліно-8-сульфонату амонію, що демонструє цілісність мембран, власна білкова флуоресценція, константа Штерна — Фольмера (швидкість гасіння вільних радикалів), мікров’язкість) [23, 25, 37].
Отримані клініко-біохімічні результати демонструють гепатопротективну дію тіотриазоліну. А з огляду на низку серйозних побічних ефектів базових засобів (антибіотиків, противірусних засобів, антиагрегантів, нестероїдних протизапальних засобів), спрямованих на порушення тонких ланок метаболізму кардіоміоцитів, ендотеліоцитів, гепатоцитів тощо, введення тіотриазоліну в комплексну терапію постковідного синдрому може підвищити безпечність пропонованого медикаментозного лікування.
Крім того, було досліджено вплив тіотриазоліну на вміст IFN-γ, IL-4, IL-2 і IL-17 у плазмі крові хворих на постковідний синдром. Група хворих, які отримували тіотриазолін, мали статистично вірогідно вищий рівень IFN-γ — в 1,72 раза порівняно з групою хворих при находженні та в 1,62 раза — порівняно з групою хворих, які отримували базисну терапію. Широко відомо, що IFN-γ є активатором мононуклеарних клітин і стимулює експресію Mac-1, підсилює ендоцитоз і фагоцитоз моноцитами, активує макрофаги. Крім того, IFN-γ стимулює диференціювання Т- і В-лімфоцитів, підсилює проліферацію активованих В-клітин і взаємодіє із IL-2, що підсилює синтез легких ланцюгів імуноглобулінів. Важливо, що IFN-γ активує нейтрофіли, NK-клітини й ендотеліальні клітини судин і знижує ризики формування дисфункції ендотелію.
Також було встановлено, що в пацієнтів з пост–ковідним синдромом при надходженні в стаціонар була зареєстрована підвищена концентрація IL-4, IL-2 і IL-17 у 8,3; 9,4 раза і на 100 % відповідно порівняно з відносно здоровими людьми, що вказує на продовження запального процесу після гострого періоду SARS-CoV-2. Низкою досліджень встановлено, що системне гіперзапалення, пов’язане з уродженим імунітетом (провокується зв’язуванням спайкового білка SARS-CoV-2 (S1) з експресуючими АПФ-2 клітинами), нейросудинною ендотеліальною дисфункцією, ушкодженням гематоенцефалічного бар’єра й активацією вродженого імунітету ЦНС, потенційно спричиняє розвиток ускладнень ЦНС, пов’язаних із SARS-CoV-2. Подальше ушкодження ендотелію периферичних судин через прямий ушкоджувальний вплив вірусної інфекції на ендотелій зумовлює ендотеліїт і пригнічення ендотеліальної NOS.
Цитокіни, що виділяються при периферичному запаленні, можуть збільшувати проникність ГЕБ, забезпечуючи шлях вірусу до проникнення в мозок. Потрапляючи до ЦНС, SARS-CoV-2 може інфікувати астроцити й мікроглію, активуючи каскад нейрозапалення й нейродегенерації за рахунок вивільнення фактора некрозу пухлини, цитокінів та інших медіаторів запалення. Прозапальні інтерлейкіни й TNF-α можуть призводити до апоптозу лімфоцитів. Активація цитокінів також може бути пов’язана з атрофією лімфоїдних органів, у тому числі селезінки, що також знижує кількість циркулюючих лімфоцитів. Нарешті, лактат-ацидоз, який є вираженим у пацієнтів у ранній постковідний період, також може інгібувати проліферацію лімфоцитів. Великомасштабні дослідження показали, що підвищені циркулюючі рівні IL-2, IL-4 і IL-17 є потенційними факторами ризику тяжкості й високої смертності при COVID-19. Більше того, сироваткові рівні IL-2, IL-4 і IL-17 були вищими в пацієнтів з тяжкою формою COVID-19 порівняно з пацієнтами з нетяжкою формою. На тлі базисної терапії з доданням до неї тіотриазоліну концентрація IL-4, IL-2 і IL-17 знижувалася відповідно на 60, 56 і 62 % порівняно з показниками при надходженні. Причому в пацієнтів на тлі призначення тіотриазоліну показники всіх інтерлейкінів статистично вірогідно перевищували показники групи пацієнтів, яким було призначено базисну терапію. Відомо, що прогресування COVID-19 до ускладненого захворювання може бути наслідком надмірної, нерегульованої імунної відповіді хазяїна й автоімунного ушкодження. Попередні дослідження показують, що інгібування цитокінового шляху на рівні ІL-4 за допомогою лікарських засобів може бути ефективним способом керування даною дисрегуляцією. Інгібування ІL-17 є важливим стратегічним аспектом запобігання респіраторному дистрес-синдрому при COVID-19. Також повідомлялося, що підвищення ІL-17 призводить до більш тяжкого постковідного міокардиту. Це говорить про те, що потенційна анти-ІL-17 терапія може відігравати роль у зниженні захворюваності й смертності, що пов’язані з міокардитом, спричиненим вірусом COVID-19.
У хворих з постковідним синдромом при надходженні спостерігались лейкопенія і лімфопенія. У постковідний період виявляються клінічні прояви захворювання — виражене системне підвищення прозапальних цитокінів, лейкопенія і лімфопенія. При надходженні в переважної більшості пацієнтів спостерігалася лімфоцитопенія (83,2 %), тоді як у 36,2 % виявлено тромбоцитопенію, а в 33,7 % — лейкопенію. У разі тяжкого перебігу захворювання ці порушення були більш вираженими порівняно з помірним перебігом захворювання (96,1 % проти 80,4 % — лімфоцитопенія; 57,7 % проти 31,6 % — тромбоцитопенія і 61,1 % проти 28,1 % — лейкопенія). Показано, що лімфоцити експресують на своїй поверхні АПФ-2, тому SARS-CoV-2 може безпосередньо інфікувати ці клітини і, зрештою, призводити до їх лізису. Лімфоцитопенія також може бути пов’язана з атрофією лімфоїдних органів, у тому числі селезінки, що також знижує кількість циркулюючих лімфоцитів. Базисна терапія з включенням до неї тіотриазоліну приводила до нормалізації досліджуваних показників. Отримані результати не суперечать раніше проведеним –доклінічним дослідженням, якими показано, що тіотриазолін знижує вміст циркулюючих імунних та імуномодулюючих комплексів, обмежує викид ними медіаторів запалення, знижує експресію прозапального цитокіну IL-1b, стабілізує мембрани, підвищує рівень інтерферону. Тіотриазолін стимулює проліферацію лімфоцитів, синтез імуноглобулінів, посилює активність Т-лімфоцитів. Також тіотриазолін гальмує АФК-залежні шляхи синтезу прозапальних простагландинів і прозапальних цитокінів.
Отже, введення в комплексну базисну терапію постковідного синдрому препарату тіотриазолін у вигляді таблеток (по 200 мг двічі на день) або супозиторіїв далмаксін (0,2 г двічі на день) протягом 30 днів приводило до вірогідного підсилення базисної ендотеліопротективної та антикоагулянтної терапії, сприяло профілактиці тромбоутворення на тлі поліпшення стану міокарда й ендотелію судин, а також зменшувало порушення гепатобіліарної системи, обумовлені як самим захворюванням, так і побічними реакціями базисної терапії.
Висновки
1. Включення в базисну комплексну терапію постковідного синдрому препарату тіотриазолін у вигляді таблеток (по 200 мг двічі на день) або супозиторіїв далмаксін (супозиторій 0,2 г двічі на день) протягом 30 діб (33 пацієнти отримували таблетки, 9 — супозиторії) приводить до вірогідного підвищення ефективності базисної антикоагулянтної та антиагрегаційної терапії і сприяє профілактиці тромбоутворення. Використання лікарської форми супозиторіїв обумовлено тим, що можуть виникати певні незручності при дотриманні пацієнтами з багатьма супутніми захворюваннями схеми прийму таблетованих лікарських засобів. Для пацієнтів у тяжкому стані такий шлях введення буде більш зручним. При цьому слід зазначити, що різниці в ефективності засобу тіотриазолін у різних лікарських формах — таблетки і супозиторії — немає (попередніми дослідженнями будо доведено високу гепатопротективну ефективність тіотриазоліну саме у вигляді такої лікарської форми, як супозиторії). Введення в базисну терапію тіотриазоліну приводить до зменшення агрегаційної активності тромбоцитів і швидкості агрегації до показників відносно здорових пацієнтів.
2. Застосування тіотриазоліну приводить до вірогідного підвищення експресії eNOS (вірогідне підвищення щодо групи пацієнтів при надходженні в середньому на 134,3 % і щодо групи базисної терапії — на 73,8 %), що свідчить про ендотеліопротективну активність досліджуваного препарату.
3. Тіотриазолін вірогідно знижує в крові хворих рівень D-димеру (вірогідне зниження щодо групи пацієнтів при надходженні на 32,3 % і щодо групи базисної терапії — на 16,1 %) (біохімічний маркер тромбоутворення), а також нормалізує показник МНВ, що відображає стан згортальної системи крові. Тіотриазолін вірогідно зменшує агрегаційну активність тромбоцитів на 32 % і швидкість агрегації на 30-ту секунду — на 54 % (щодо показників відносно здорових пацієнтів). Усе це свідчить про виражені антиагрегантні й фібринолітичні ефекти тіотриазоліну, а також про його здатність знижувати ризики інфарктів та інсультів при постковідному синдромі.
4. Введення в базисну терапію хворих з постковідним синдромом тіотриазоліну у вигляді таблеток або супозиторіїв дає гепатопротективний ефект, що проявляється в зниженні АЛТ і АСТ на 56,4 і 67,1 % порівняно з показниками групи хворих при надходженні і на 48,5 і 61,0 % порівняно з групою хворих, які отримували базисну терапію; у нормалізації тимолової проби, ГГТ і значень загального білірубіну й зменшенні прояву цитолітичного синдрому. Важливо зазначити, що в групі хворих, які отримували базисну терапію, значення АСТ залишились підвищеними. Також у пацієнтів основної групи нормалізувалась білоксинтезуюча функція печінки, про що свідчила нормалізація показника тимолової проби на відміну від групи хворих, які отримували базисну терапію.
5. У 40 хворих з 42 (95,2 %), які отримували курсом тіотриазолін, відмічалась позитивна динаміка, а саме зменшення астеновегетативного, холестатичного, больового синдрому в епігастрії і правому підребер’ї. У групі, яка отримувала базову терапію, позитивна динаміка мала місце тільки в 9 хворих із 20 (45 %). З огляду на низку серйозних побічних ефектів базових засобів, спрямованих на порушення тонких ланок метаболізму кардіоміоцитів, ендотеліоцитів, гепатоцитів тощо, введення в комплексну терапію постковідного синдрому тіотриазоліну підвищує безпеку базової медикаментозної терапії.
6. Призначення тіотриазоліну привело до вірогідного поліпшення загальноклінічних показників у пацієнтів з постковідним синдромом — зникли скарги на серцебиття, артеріальний тиск стабілізувався (без додаткової корекції гіпотензивними препаратами), зникли слабкість і підвищена втома. Сатурація в 40 (95,2 %) пацієнтів основної групи підвищилася до 98–99 %. У контрольній групі тільки в 9 (45 %) з 20 пацієнтів сатурація була на рівні 98 %.
7. Призначення тіотриазоліну приводить до вірогідного поліпшення показників імунної системи в пацієнтів із постковідним синдромом — вірогідно підвищувався рівень інтерферону (в 1,62 раза порівняно з групою хворих, які отримували базисну терапію) і знижувалась концентрація IL-4, IL-2 і IL-17 на 60, 56 і 62 % відповідно порівняно з показниками хворих при надходженні. Також тіотриазолін приводив до нормалізації показників периферичної крові — зменшення лейкопенії та лімфопенії.
Список литературы
1. Chary M.A., Barbuto A.F., Izadmehr S., Hayes B.D., Burns M.M. COVID-19: Therapeutics and Their Toxicities. J. Med Toxicol. 2020 Jul. 16(3). 284-294. Published online 2020 Apr 30. doi: 10.1007/s13181-020-00777-5.
2. Fan E., Beitler J.R., Brochard L., Calfee C.S., Ferguson N.D., Slutsky A.S., Brodie D. COVID-19-associated acute respiratory di–stress syndrome: is a different approach to management warranted? Lancet Respir. Med. 2020 Aug. 8(8). 816-821. doi: 10.1016/S2213-2600(20)30304-0.
3. Wilson J.G., Simpson L.J., Ferreira A.M., Rustagi A., Roque J., Asuni A. et al. Cytokine profile in plasma of severe –COVID-19 does not differ from ARDS and sepsis. JCI Insight. 2020 Sep 3. 5(17). e140289. doi: 10.1172/jci.insight.140289. PMID: 32706339; PMCID: PMC7526438.
4. Zhao Z., Xie J., Yin M. et al. Clinical and Laboratory Profiles of 75 Hospitalized Patients with Novel Coronavirus Disease 2019 in Hefei, China. MedRxiv. 2020. https://doi.org/10.1101/2020.03.01.20029785.
5. Barnes B.J., Adrover J.M., Baxter-Stoltzfus A. et al. Targe–ting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020. Vol. 217. № 6. Art. ID e20200652.
6. Smeitink J., Jiang X., Pecheritsyna S. et al. Hypothesis: mPGES-1-derived Prostaglandin E2, a so far missing link in –COVID-19 pathophysiology? Preprints. 2020. 2020040180.
7. Conti P., Ronconi G., Caraffa A., Gallenga C.E., Ross R., Frydas I., Kritas S.K. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J. Biol. Regul. Homeost. Agents. 2020 March-April. 34(2). 327-331. doi: 10.23812/CONTI-E. PMID: 32171193.
8. Landi L., Ravaglia C., Russo E. et al. Blockage of interleukin-1β with canakinumab in patients with Covid-19. Sci Rep. 2020. 10. 21775. https://doi.org/10.1038/s41598-020-78492-y.
9. Guan S.P., Seet R.C.S., Kennedy B.K. Does eNOS derived nitric oxide protect the young from severe COVID-19 complications? Ageing Res. Rev. 2020. 64. 101201. doi:10.1016/j.arr.2020.101201.
10. Green S.J. Covid-19 accelerates endothelial dysfunction and nitric oxide deficiency. Microbes Infect. 2020. 22 (4–5). 149-150. doi: 10.1016/j.micinf.2020.05.006.
11. Varga Z., Flammer A.J., Steiger P. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020. pii: S0140-6736(20)30937-5. https://doi.org/10.1016/S0140-6736(20)30937-5.
12. Lapenna D. Antioxidant therapy in COVID-19: The crucial role of early treatment and antioxidant typology. Clinical Infectious Diseases. 2021. ciab055. https://doi.org/10.1093/cid/ciab055.
13. Fratta Pasini A.M., Stranieri C., Cominacini L., Mozzini C. Potential Role of Antioxidant and Anti-Inflammatory Therapies to Prevent Severe SARS-Cov-2 Complications. Antioxidants (Basel). 2021. 10(2). 272. Published 2021 Feb 10. doi: 10.3390/antiox10020272.
14. Impact of Thiol-Disulfide Balance on the Binding of –Covid-19 Spike Protein with Angiotensin-Converting Enzyme 2 Receptor Sanchita Hati and Sudeep Bhattacharyya ACS Omega. 2020. 5(26). 16292-16298. DOI: 10.1021/acsomega.0c02125.
15. Miller B., Silverstein A., Flores M. et al. Host mitochondrial transcriptome response to SARS-CoV-2 in multiple cell models and clinical samples. Sci Rep. 2021. 11. 3. https://doi.org/10.1038/s41598-020-79552-z.
16. Chernyak B.V., Popova E.N., Prikhodko A.S., Grebenchi–kov O.A., Zinovkina L.A., Zinovkin R.A. COVID-19 and Oxidative Stress. Biochemistry. 2020. 85(12) 1543-1553. Published online 2020 Dec 28. doi: 10.1134/S0006297920120068.
17. Velavan T.P., Meyer C.G. Mild versus severe COVID-19: Laboratory markers. Int. J. Infect. Dis. 2020 Jun. 95. 304-307. Published online 2020 Apr 25. doi: 10.1016/j.ijid.2020.04.061.
18. Smith M., Smith J.C. Repurposing Therapeutics for –COVID-19: Supercomputer-Based Docking to the SARS-CoV-2 Viral Spike Protein and Viral Spike Protein-Human ACE2 Interface. ChemRxiv. 2020. Preprint.
19. Iqubal А., Iqubal M.K., Hoda F., Najmi A.K., Haque S.E. COVID-19 and cardiovascular complications: an update from the underlying mechanism to consequences and possible clinical intervention. Expert Rev. Anti Infect. Ther. 2021. 1–10. Published online 2021 Mar 5. doi: 10.1080/14787210.2021.1893692.
20. Suhail S., Zajac J., Fossum C. et al. Role of Oxidative Stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) Infection: A Review. Protein J. 2020. 39. 644-656. https://doi.org/10.1007/s10930-020-09935-8.
21. Tyagi S.C., Singh М. Multi-organ damage by COVID-19: congestive (cardio-pulmonary) heart failure, and blood-heart barrier leakage. Mol. Cell. Biochem. 2021 Jan 22. 1-5. doi: 10.1007/s11010-021-04054-z [Epub ahead of print].
22. Yeshun Wu, Xiaolin Xu, Zijun Chen, Jiahao Duan, Kenji Hashimoto, Ling Yang, Cunming Liu, Chun Yang. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immun. 2020 Jul. 87. 18-22. Published online 2020 Mar 30. doi: 10.1016/j.bbi.2020.03.031.
23. Беленичев И.Ф., Бухтиярова Н.В., Черний В.И., Павлов С.В., Колесник Ю.М., Абрамов А.А. Рациональная нейропротекция. Донецк: Издатель Заславский А.Ю., 2009. 262 с.
24. Бєленічев І.Ф., Безруков В.В., Купраш Л.П., Горчакова Н.О., Нагорна О.О., Гріненко Ю.О. та ін. Фармакотерапія в геріатричній клініці. Дніпро: Журфонд, 2020. 166 с.
25. Беленичев И.Ф., Мазур И.А., Волошин Н.А., Визир В.А. Метаболические кардиопротекторы: фармакологические свойства и применение в клинической практике: Метод. реком. Запоріжжя — Київ: МОЗ України, 2007. 177 с.
26. Мазур И.А., Чекман И.С., Беленичев И.Ф., Горчакова Н.А., Волошин Н.А., Кучеренко Л.И. Метаболитотропные препараты. Запорожье, 2007. 304 с.
27. Беленичев И.Ф., Визир В.А., Мамчур В.И. Место тио–триазолина в галерее современных метаболитотропных лекарственных средств. Запорожский медицинский журнал. 2019. Т. 21. № 1. С. 119-128.
28. Мазур И.А., Волошин Н.А., Кучеренко Л.И., Беленичев И.Ф., Визир В.А. Тиотриазолин, тиодарон в лечении сердечно-сосудистой патологии. Запорожье: Печатный Мир, 2011. 305 с.
29. Беленичев И.Ф., Нагорная Е.А., Горбачева С.В., Горчакова Н.А., Бухтиярова Н.В. Тиол-дисульфидная система: роль в эндогенной цито- и органопротекции, пути фармакологической модуляции. Киев: Юстон, 2020. 232 с.
30. Бєленічев І.Ф., Резниченко Ю.Г., Резниченко Н.Ю., Риженко О.І. Перинатальні ураження нервової системи. Запоріжжя: Просвіта, 2020. 364 с.
31. Barker-Davies R., O’Sullivan O., Senaratne K. et al. The Stanford Hall consensus statement for post-COVID-19 rehabilitation. Br. J. Sports Med. 2020. 54. 949-959.
32. Hasichaolu Z.X., Li X. et al. Circulating cytokines and lymphocyte subsets in patients who have recovered from COVID-19. Biomed. Res. Int. 2020 Nov 26. 2020. 7570981.
33. Zhang W., Zhao Y., Zhang F. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): the perspectives of clinical immunologists from China. Clinical Immunology. 2020. 214. 108393
34. Hu H., Pan H., Li R., He K., Zhang H., Liu L. Increased Circulating Cytokines Have a Role in COVID-19 Severity and Death With a More Pronounced Effect in Males: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2022. 13. 802228. doi: 10.3389/fphar.2022.802228
35. Pacha O., Sallman M.A., Evans S.E. COVID-19a: a case for inhibiting IL-17? Nat. Rev. Immunol. 2020 Jun. 20(6). 345-346. doi: 10.1038/s41577-020-0328-z. PMID: 32358580; PMCID: PMC7194244.
36. Belenichev I., Kucherenko L., Pavlov S., Bukhtiyarova N., Popazova O., Derevianko N., Nimenko G. Therapy of post-COVID-19 syndrome: improving the efficiency and safety of basic metabolic drug treatment with tiazotic acid (thiotriazoline). Pharmacia. 2022. 69(2). 509-516. https://doi.org/10.3897/pharmacia.69.e82596.
37. Інформаційний лист «Підвищення ефективності комплексного лікування хворих з постковідним синдромом». Київ, 2021.
38. Інформаційний лист «Оптимізація імунореабілітації при коронавірусній інфекції COVID-19». Київ, 2022.