Резюме
Хвороба Шарко — Марі — Тута, або невральна аміотрофія, ще відома як спадкова моторно-сенсорна невропатія. Це генетично детерміноване захворювання, яке має різні варіанти успадкування, основу якого становлять розлади нервової та опорно-рухової системи. Дебют захворювання можливий як в дитячому, так і в зрілому віці. Вченими було ідентифіковано та клінічно описано понад тридцять генетичних мутацій, які формують індивідуальний унікальний паспорт порушень послідовності групи органічних сполук або нуклеотидів у молекулах нуклеїнових кислот. Розрізняють автосомно-домінантний, автосомно-рецесивний та зчеплений зі статтю типи генетичного наслідування хвороби Шарко — Марі — Тута. Захворювання вперше описане 1886 р. та зустрічається з частотою від 10 до 40 на 100 тис. населення. В Україні зареєстровано більш ніж 15 тисяч випадків хвороби Шарко — Марі — Тута. Слід зазначити, що білок 22 (PMP22) став першим описаним геном, що спричинює хворобу Шарко — Марі — Тута. Автосомно-домінантний тип Шарко — Марі — Тута 1 є найпоширенішою формою захворювання. При огляді пацієнтів з цією формою невральної аміотрофії невролог звертає увагу на порушення ходьби різного ступеня тяжкості (степаж), деформацію стоп (pes cavus) з проявами дистальної гіпотрофії, зниження або відсутність сухожилкових і періостальних рефлексів, слабкість і втрату чутливості в дистальних відділах кінцівок. Гіпотрофічні зміни у кінцівках мають безперервно прогредієнтний характер. Внаслідок атрофії м’язів та деформації стоп формується високе склепіння та екстензія великого пальця (стопа Фрідрейха). На сьогодні не існує патогенетично обґрунтованого лікування хвороби Шарко — Марі — Тута, але багатовекторний підхід щодо комплексних реабілітаційних заходів створює необхідні умови для ефективного стримування проявів неминучого прогресування цієї патології.
Charcot-Marie-Tooth disease is also called hereditary motor and sensory neuropathy. It is a genetically determined disease that has different variants of inheritance whose substratum is neuromuscular disorders. It may begin during childhood or later in life. Scientists have identified and described more than thirty genetic mutations that form an individual unique passport of violations of the sequence of a group of organic compounds or nucleotides in nucleic acid molecules. There are autosomal dominant, autosomal recessive and X-linked types of genetic inheritance of Charcot-Marie-Tooth disease. This pathology was first described in 1886 and affects about 10 to 40 per 100,000 population. More than 15,000 cases of Charcot-Marie-Tooth disease have been registered in Ukraine. It should be noted that peripheral myelin protein 22 became the first described gene causing Charcot-Marie-Tooth disease. Autosomal dominant Charcot-Marie-Tooth type 1 is the most common form of the disease. When observing patients, a neurologist pays attention to gait disturbances (steppage gait), foot distortion (pes cavus), distal hypotrophy, decrease or absence of tendon and periosteal reflexes, weakness and loss of sensitivity in the distal parts of the limbs. Hypotrophic changes in the limbs are continuously progressive. As a result of muscle atrophy and foot deformity, high-arched feet and extension of the big toe (Friedreich’s foot) are formed. To date, no pathogenetically substantiated treatment exists for Charcot-Marie-Tooth disease, but a multi-vector approach to comprehensive rehabilitation measures creates the necessary conditions to effectively control the manifestations of the inevitable progression of this pathology.
Хвороба Шарко — Марі — Тута, або невральна аміотрофія, відома як спадкова моторно-сенсорна невропатія, що охоплює клінічно та генетично гетерогенну групу розладів, основними проявами яких є м’язова слабкість, дистальні м’язові гіпотрофії, втрата чутливості за поліневритичним типом й деформація стоп у вигляді стопи Фрідрейха. Дебют захворювання можливий як у дитячому, так і в зрілому віці.
Захворювання вперше описане 1886 р. та зустрічається з частотою від 10 до 40 на 100 тис. населення. В Україні зареєстровано більш ніж 15 тисяч випадків хвороби Шарко — Марі — Тута.
Процес дослідження патогенетичних механізмів невральної аміотрофії Шарко — Марі — Тута умовно можна розділити на два періоди. Перший — догенетичний. На цьому етапі була визначена та ретельно досліджена зростаюча кількість клінічних випадків, створена класифікація захворювання, виділено два основні її типи: Шарко — Марі — Тута 1 (демієлінізуючий) і Шарко — Марі — Тута 2 (аксональний).
Другий період вивчення невральної аміотрофії розпочався близько тридцяти років тому й був пов’язаний з генетичною ідентифікацією подвоєної хромосоми 17, яка містить периферичний мієліновий білок 22 (PMP22). Зазначимо, що білок 22 (PMP22) став першим описаним геном, що спричинює хворобу Шарко — Марі — Тута.
На сьогодні виявлено понад 30 генів, порушення функціонування яких зумовлює розвиток захворювання, а генетичний скринінг створює необхідні умови для встановлення точного діагнозу.
Зауважимо, що автосомно-домінантний тип Шарко — Марі — Тута 1 є найпоширенішою формою захворювання.
Як правило, при огляді пацієнтів з невральною аміотрофією невролог звертає увагу на порушення ходьби різного ступеня тяжкості (степаж), деформацію стоп (pes cavus) з проявами дистальної гіпотрофії, зниження або відсутність сухожилкових і періостальних рефлексів, слабкість і втрату чутливості в дистальних відділах кінцівок. Слід зазначити, що гіпотрофічні зміни у кінцівках мають безперервно прогредієнтний характер. Атрофії спочатку зачіпають литкові м’язи, а з часом патологічний процес поширюється на проксимальні відділи нижніх і верхніх кінцівок. Особливості розвитку гіпотрофії м’язів нижніх кінцівок характеризуються деякою аналогією з «ногами лелеки» або «перевернутою пляшкою».
Унаслідок атрофії м’язів та деформації стоп формується високе склепіння та екстензія великого пальця (стопа Фрідрейха). Відмічається зниження чутливості за поліневритичним типом. Перебіг хвороби повільно прогресуючий. Пацієнти часто скаржаться на появу болю чи парестезій у кінцівках. У міру прогресування захворювання виявляється зниження слуху та зору, у тяжких випадках нерідко відмічаються ознаки дихальної недостатності через порушення функції діафрагмального м’яза.
Підкреслимо, що, як правило, невральна аміотрофія Шарко — Марі — Тута не корелює з тривалістю життя хворого, але безумовно впливає на його якість, нерідко суттєво інвалідизуючи пацієнта на пізніх стадіях захворювання.
На сьогодні не існує патогенетично обґрунтованого лікування хвороби Шарко — Марі — Тута, але багатовекторний підхід щодо комплексних реабілітаційних заходів створює необхідні умови для ефективного стримування проявів неминучого прогресування цієї патології.
Нижче наведено власне клінічне спостереження пацієнта з клінічними ознаками невральної аміотрофії Шарко — Марі — Тута.
Хворий М., 53 роки, звернувся на консультацію до невролога клініки «Аксімед» зі скаргами на слабкість у ногах, більше в правій, труднощі з підійманням правої стопи, періодичний біль у поперековій ділянці хребта.
З анамнезу хвороби відомо, що з дитинства у пацієнта були стопи з високим склепінням. У віці 20 років почав відмічати слабкість у ногах, що супроводжувалась певним розладом ходьби. Ці порушення збільшувались упродовж життя.
Сімейний анамнез обтяжений, відомо, що рідний брат та батько пацієнта теж мають деформацію стоп та їх високе склепіння.
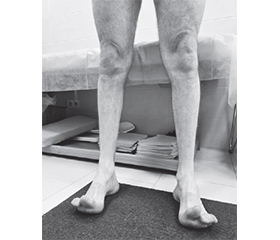
У процесі дослідження неврологічного статусу було відмічено наступне: пацієнт астенізований, емоційно лабільний, черепні нерви без змін, очні щілини S = D, зіниці S = D, фотореакції зіниць збережені, конвергенція не порушена, рухи очних яблук у повному обсязі. Ністагму немає. Лице симетричне. Язик розташований по середній лінії. Мовлення та ковтання не порушені. Сухожилкові та періостальні рефлекси з верхніх кінцівок не змінені, з нижніх — не викликаються. Наявна виражена гіпотрофія м’язів гомілок та стоп. Деформація стоп за типом стопи Фрідрейха. Хода порушена, відмічається степаж. Стояння на пальцях значно утруднене, на п’ятках — неможливе. Зниження сили в розгиначах правої стопи за шкалою м’язової слабкості відповідає 2 балам, а в згиначах — 3 балам. Координаторні проби виконує впевнено. Глибока чутливість збережена. Поверхнева гіпестезія у нижніх кінцівках за типом «шкарпеток». Патологічні рефлекси не викликаються. Функції органів малого таза не порушені.
Виходячи з вимог діагностичного протоколу пацієнту було виконано: стимуляційну електронейроміографію (ознаки грубого аксонально-демієлінізуючого ураження сенсорних та моторних волокон нервів нижніх кінцівок за поліневритичним типом зі зниженням амплітуди М-відповідей); МРТ попереково-крижового відділу хребта (ознаки дегенеративно-дистрофічних змін попереково-крижового відділу хребта, ускладнених протрузіями міжхребцевих дисків L2-S1). Також отримані результати загальноклінічного обстеження, включно з оглядом терапевта, офтальмолога, ортопеда, проаналізовані дані лабораторних досліджень. Пацієнту запропоновано генетичний скринінг.
З урахуванням вищенаведеного хворому встановлений клінічний діагноз: невральна аміотрофія Шарко — Марі — Тута з помірним дистальним парапарезом нижніх кінцівок й порушенням функції ходьби.
На фоні проведених комплексних реабілітаційних заходів, які включали кінезіотерапію, апаратні та фізіотерапевтичні методи відновлювальної терапії, рефлексотерапію, лікувальний масаж, пацієнт відмітив поліпшення ходи, координації рухів та загальної активності. Також був розроблений індивідуальний план локомоторної терапії в умовах амбулаторного диспансерного нагляду.
Конфлікт інтересів. Автори заявляють про відсутність конфлікту інтересів та власної фінансової зацікавленості при підготовці даної статті.
Отримано/Received 06.07.2024
Рецензовано/Revised 17.07.2024
Прийнято до друку/Accepted 26.07.2024
Список литературы
1. Тріщинська М.А., Дубинецька В.М. Невральна аміотрофія Шарко — Марі — Тута (клінічний випадок). Міжнародний неврологічний журнал. 2023. Т. 19. № 6.
2. Сиделковський А.Л. Нейрореабилитация. Основы теории и практики. Киев, 2022. 592 с.
3. Сіделковський О., Овсянніков О., Марусіченко В. Діагностичні шкали і тести в неврології, нейрохірургії і нейрореабілітації. Київ: Пабліш Про, 2022. 296 с.
4. Klein C.J. Charcot-Marie-Tooth Disease and Other Hereditary Neuropathies. Peripheral Nerve and Motor Neuron Disorders. October 2020. Vol. 26. Р. 1224-1256. doi: 10.1212/CON.0000000000000927.
5. Reilly M., Murphy S. et al. Charcot-Marie-Tooth disease. Journal of the Peripheral Nervous System. 2011. Vol. 16. Р. 1-14.
6. Okamoto Y., Takashima H. The Current State of Charcot-Marie-Tooth Disease Treatment. Genes. 2023. Vol. 14(7). Р. 1391. doi.org/10.3390/genes14071391.
7. Stavrou M., Sargiannidou I., Georgiou E. еt al. Therapies for Charcot-Marie-Tooth Inherited Neuropathies. Int. J. Mol. Sci. 2021. Vol. 22. Р. 6048.
8. Pareyson D., Saveri P., Pisciotta C. New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr. Opin. Neurol. 2017. Vol. 30. Р. 471-480.

/65.jpg)