Епілептичні напади виникають у всіх пацієнтів, хворих на епілепсію, але не всі пацієнти, які мали епілептичний напад, страждають на епілепсію. До 40 % усіх епілептичних нападів виникають у людей з гострим ураженням головного мозку, але це ще не означає, що у них є епілепсія.
Слід зазначити, що в англійській мові гострий симптоматичний напад (ГСН) — це acute symptomatic seizure, де слово seizure означає «епілептичний напад», що не відображається в українському перекладі. Отже, вживаючи термін «гострий симптоматичний напад», ми апріорі маємо на увазі, що він епілептичний.
ГСН «виникає в тісному часовому зв’язку з гострим ураженням ЦНС, яке може бути метаболічним, токсичним, структурним, інфекційним або внаслідок запалення» [1].
У клінічній практиці часто використовується кілька термінів, схожих на термін «гострий симптоматичний напад», наприклад такі, як «спровокований напад», «ситуативний напад» та «реактивний напад». Міжнародна протиепілептична ліга (МПЕЛ) запропонувала, щоб ці терміни були синонімами гострого симптоматичного нападу [1].
У дорослих найпоширенішою етіологією ГСН є цереброваскулярні захворювання, черепно-мозкова травма (ЧМТ), абстиненція від наркотиків і алкоголю та інфекції ЦНС, причому на кожну етіологію припадає приблизно однакова частка випадків. Менш поширеними причинами ГСН є метаболічні розлади, специфічні енцефалопатії, інтоксикації та еклампсія [1].
Гострий симптоматичний напад виникає під час системного ураження головного мозку або в тісному часовому зв’язку із задокументованим ураженням мозку [2–5]. Це визначення було створено для використання в епідеміологічних дослідженнях. У класифікації епілепсій та епілептичних синдромів (Комісія з класифікації та термінології МПЕЛ, 1989) гострі симптоматичні напади класифікуються як напади, пов’язані із ситуацією. Ця категорія також охоплює напади, для яких немає очевидної причини. МПЕЛ у 2005 р. визначила епілепсію як «розлад, що характеризується стійкою схильністю до епілептичних нападів і нейробіологічними, когнітивними, психологічними та соціальними наслідками цього стану. Таке визначення потребує наявності принаймні одного епілептичного нападу» [6].
ГСН відрізняються від епілепсії кількома важливими аспектами. По-перше, на відміну від епілепсії, безпосередню причину цих нападів можна чітко визначити настільки, наскільки можна бути впевненим у причинно-наслідковому зв’язку. Близька часова послідовність свідчить про наявність причинно-наслідкового зв’язку ГСН з такими станами, як травма голови або інсульт, коли вони безпосередньо передують нападу або виникають одночасно з ним. Біологічна імовірність також підтверджує причинність, як це видно на прикладі гострого порушення цілісності мозку або метаболічного гомеостазу у зв’язку з пошкодженням. Часто існує «ефект дози», коли більш серйозні травми призводять до більш високого ризику нападів. Хоча коефіцієнти ризику не були розраховані, вони, ймовірно, величезні. По-друге, на відміну від епілепсії, існує залежність проявів ГСН від схильності до нападів. Хоча люди, які мають певні пошкодження головного мозку, іноді мають підвищений ризик розвитку епілепсії в майбутньому, гострі симптоматичні судоми навряд чи повторяться, якщо не повториться основний гострий причинний стан [7]. Як наслідок, більшість людей не потребує тривалого лікування протинападовими препаратами (ПНП), хоча таке лікування може бути виправданим на короткостроковій основі, доки не мине гостра фаза.
Люди з епілепсією також можуть мати ГСН. Питання про те, чи є у них більша ймовірність виникнення такого епізоду у зв’язку з конкретною ситуацією, — це питання для майбутнього вивчення. На думку авторів цього огляду та більшості закордонних експертів, синонімами гострих симптоматичних нападів є спровоковані та ситуаційні епілептичні напади у пацієнтів з верифікованою епілепсією. Депривація сну недостатньо вивчена як провокатор нападів у людей без епілепсії або як фактор ризику виникнення нападів у людей з епілепсією. Хоча критерії для визначення ГСН є дещо умовними, це перша спроба надати коди рівням ризику, пов’язаним із широким спектром ушкоджень головного мозку.
При цьому слід відзначити, що рефлекторна епілепсія та світлочутлива епілепсія не є гострими симптоматичними нападами, а є саме епілепсіями, що визначаються реакцією на специфічний подразник.
Кілька епідеміологічних досліджень повідомляють про частоту ГСН. Такі напади становлять в середньому 34 % усіх афебрильних нападів [5]. Додавання фебрильних судом збільшує цю кількість приблизно до 55 % усіх нападів [5]. Значні відмінності в частці людей з ГСН в дослідженнях можуть спричинюватись труднощами у їх визначенні. Такі напади рідко вказуються як діагноз. Найчастіше діагноз основного захворювання закодований, що робить дослідження, які покладаються на коди нападів за МКХ, неефективними через імовірний грубий недолік. Крім того, цих осіб рідко направляють на довготривале спостереження до неврологів. З огляду на гострий характер основного ураження електроенцефалографічне обстеження виконується далеко не у всіх пацієнтів, навіть якщо воно є доречним, а його результати не завжди є інформативними. Таким чином, ці джерела для ідентифікації випадку є ненадійними. У дослідженнях, які спираються на опитування, необхідний досить складний інструментарій, щоб відрізнити ГСН від неспровокованих нападів. Таким чином, хоча причинно-наслідковий зв’язок і прогноз гострих симптоматичних нападів відрізняються від епілепсії, деякі епідеміологічні дослідження могли включати такі напади як епілепсію [8] або не змогли відрізнити їх від неспровокованих нападів [9].
ГСН відрізняються від неспровокованих нападів кількома аспектами, тому не входять у визначення епілепсії. По-перше, на відміну від неспровокованих нападів, завжди має існувати чітко ідентифікований супутній гострий причинний стан, який виник ближче до моменту нападу. Причиною може бути гостре порушення структурної цілісності головного мозку, таке як крововилив у кору головного мозку, або порушення функції мозку внаслідок, зокрема, алкогольної абстиненції, наявної під час нападу. По-друге, гострі симптоматичні судоми зазвичай не повторюються після того, як провокуючий фактор або стан був усунутий або повернутий назад, а функціональна цілісність ЦНС була відновлена. ГСН відрізняються від епілепсії, за якої напади можуть повторюватися. Відсутність «стійкої схильності» після ГСН означає, що підстав для діагностики епілепсії не існує. Наприклад, якщо у пацієнта виникли два напади епілепсії через тяжку гіпонатріємію, то після її зникнення «стійкої схильності» немає. Однак відмінність між ГСН та епілепсією складніша в осіб з гострими симптоматичними нападами внаслідок деструктивних патологій мозку, таких як інсульт або травма голови, оскільки вони мають підвищений ризик розвитку епілепсії в подальшому. Те, що вважається тісним тимчасовим зв’язком між ураженням ЦНС і нападом, залежить від основної патології. Наприклад, судомний напад вважається гострим симптоматичним, якщо він виникає протягом перших семи днів після інсульту або черепно-мозкової травми [1, 10]. В інших випадках ГСН може виникнути через понад тиждень після початку ураження головного мозку за умови, що є докази тривалого активного захворювання мозку. Прикладом є гостре запальне захворювання ЦНС (наприклад, інфекційний або автоімунний енцефаліт). За інших умов потрібен більш тісний часовий зв’язок, щоб довести вірогідну причинність. Ідеться про такі розлади, як гіпонатріємія, коли мають бути ознаки низького рівня натрію в сироватці крові протягом 24 годин після нападу. Напади, які є проявом нейродегенеративного захворювання, як-от деменція Альцгеймера, можна назвати прогресуючими симптоматичними нападами. Вони не є гострими симптоматичними нападами, оскільки причина нападу не є ні тимчасовою, ні оборотною, а є стійким і прогресуючим станом. У таких випадках діагноз епілепсії можна остаточно встановити після другого нападу. Однак можна поставити відповідний діагноз навіть після першого нападу, якщо є докази того, що ризик рецидиву перевищує 60 %. Подібним чином напади, які виникають внаслідок появи більшості пухлин головного мозку, є прогресуючими симптоматичними нападами, якщо пухлина мозку не може бути повністю видалена і напади не зникають. Наприклад, у пацієнта, у якого перший напад є першим проявом менінгіоми, судомний напад можна оцінити як гострий симптоматичний, якщо пухлина повністю видалена і подальших нападів немає. Тут гостра симптоматика може бути визначена лише в ретроспективі. При розсіяному склерозі судомний напад слід розглядати як гострий симптоматичний, якщо він виникає під час або протягом семи днів після рецидиву [1, 10].
Основна проблема у визначенні ГСН виникає через складність об’єднання в одному понятті як нападів, викликаних гострими структурними патологіями мозку, так і нападів, викликаних провокуючими факторами. Є думка, що напади, викликані гострим структурним ураженням головного мозку, таким як інсульт, не повинні прирівнюватися одним терміном до нападів, спровокованих справді оборотним фактором, як-от гіпонатріємія [11]. Встановлено, що 10-річний ризик рецидиву неспровокованого судомного нападу після гострого симптоматичного нападу внаслідок інсульту становить 33 % [12]. Це суттєво, але все ж означає, що сам ГСН не кваліфікується як епілепсія, однак якщо вказати підтип інсульту, то ризик ще вищий (наприклад, згідно з прогнозом SeLECT) [13]. З іншого боку, передбачається, що гострі симптоматичні напади, спричинені оборотним фактором або станом, таким як інтоксикація або гіпонатріємія, пов’язані з дуже низьким ризиком подальших неспровокованих нападів, хоча точні дані щодо ризику рецидиву відсутні.
Різниця між спровокованим і неспровокованим нападом може бути складною для ідентифікації. Важко повністю виключити провокуючий фактор, навіть якщо судомний напад виглядає неспровокованим. З іншого боку, наявність потенційно провокаційного фактора не виключає існування основної схильності до виникнення епілептичних нападів [6].
У деяких ситуаціях, таких як виникнення нападу в безпосередньому контексті відміни алкоголю або гіпонатріємії, напад буде впевнено оцінюватися як спровокований і не призведе до встановлення діагнозу епілепсії. У контексті депривації сну ситуація менш зрозуміла. Тривала депривація сну потенційно може спровокувати напад у людини без будь-якої основної схильності до розвитку нападів, однак депривація сну також є типовим провокуючим фактором ідіопатичної генералізованої епілепсії.
Крім того, виняткова наявність спровокованих нападів не означає, що епілепсії не існує, адже, як зазначалося вище, провокуючі фактори можуть бути присутніми при кожному нападі у людей з рефлекторною епілепсією. Тут є тривала аномальна зміна функції мозку, що відповідає принаймні концептуальному визначенню епілепсії [3].
Визначення тісного тимчасового зв’язку, а також запропоновані граничні рівні лабораторних значень для гострих симптоматичних нападів через метаболічні порушення викликали критику через те, що вони є відносно довільними та не підкріплені чіткими даними [11]. Наприклад, при електролітних порушеннях гострота зміни є більш важливою для ризику нападів, ніж зміна абсолютних рівнів [14]. Якщо є підозра, що судомний напад викликаний метаболічним розладом, але порогові рівні ILAE лабораторних значень не досягаються, рекомендується не маркувати напад як гострий симптоматичний [1, 10]. Однак це не означає, що судомний напад можна назвати неспровокованим. У такому разі зв’язок із метаболічним розладом слід розглядати як невідомий, а судомний напад не слід оцінювати як епілепсію.
Гострі симптоматичні напади та смертність
Пацієнти, які страждають від гострих симптоматичних нападів, мають високий ризик смертності протягом кількох тижнів після події. Дослідження двох незалежних когорт пацієнтів із ГСН визначило летальність у 20 % у перші 30 днів після нападу [15]. Ризик смерті був значно вищим у літніх людей (старше 65 років), ніж у молодих. Цереброваскулярні захворювання та гіпоксична (аноксична) енцефалопатія були визначені як переважні причини гострих симптоматичних нападів у тих пацієнтів, у яких був летальний результат у перші 30 днів після нападу.
У дослідженні за участю госпіталізованих пацієнтів ті, у кого напади трапилися вперше, а більшість із них відбулася через гостру симптоматичну причину, мали значно вищі шанси на негативний результат (смерть або виписку до хоспісу), ніж пацієнти, які вже мали судомні напади до госпіталізації [16]. Встановлено, що цереброваскулярна хвороба є найчастішою етіологією нападів у цих пацієнтів. За нею слідують метаболічні порушення та пухлини головного мозку.
В іншому дослідженні порівнювали смертність тих пацієнтів, які мали ГСН, і тих, які мали перший неспровокований напад. У пацієнтів з гострими симптоматичними нападами рівень смертності в перші 30 днів після нападу був у 8,9 раза вищим [12]. Через 10 років спостереження між цими двома групами не було очевидної різниці за смертністю.
Смертність була особливо висока у пацієнтів з гострим симптоматичним епілептичним статусом. У дослідженні за участю 184 пацієнтів як з гострим симптоматичним, так і з неспровокованим епілептичним статусом 89 % смертей (n = 38) у перші 30 днів після події сталися власне у пацієнтів з гострим симптоматичним епілептичним статусом [17]. Смертність у пацієнтів з гострим симптоматичним епілептичним статусом становила 34 %, а у пацієнтів з неспровокованим епілептичним статусом — 5 %. Вік старше 65 років і чоловіча стать значною мірою асоціювалися з вищим ризиком смертності, і більшість пацієнтів, які померли після гострого симптоматичного епілептичного статусу, страждала від цереброваскулярних захворювань або гіпоксичної енцефалопатії. Гострий симптоматичний епілептичний статус також пов’язаний з підвищеним ризиком смертності [18].
Висока смертність у пацієнтів з гострими симптоматичними судомними нападами в основному пояснюється основною причинною патологією головного мозку [15]. Добре відомо, що етіологія таких гострих симптоматичних нападів, як цереброваскулярна хвороба, пухлини головного мозку або гіпоксична енцефалопатія, мають високий ризик смертності незалежно від того, виникають вони в поєднанні з нападом чи ні. Вплив ГСН на клінічні результати пацієнтів із гострими захворюваннями головного мозку неясний. Виникнення ГСН може просто відображати тяжкість основного гострого ураження головного мозку.
Обговорювалося питання про те, чи гострі симптоматичні напади самі по собі негативно впливають на клінічний результат пацієнта. Це питання особливо вивчалося у пацієнтів з інсультом, і деякі дослідження продемонстрували, що гострі симптоматичні напади незалежно пов’язані з вищою смертністю [19], тоді як інші не продемонстрували такого ефекту [20]. Проте недавній великий ретроспективний аналіз 1787 пацієнтів із гострими симптоматичними нападами на тлі гострого ішемічного інсульту контролювався за тяжкістю інсульту за допомогою шкали інсульту Національного інституту здоров’я (NIHSS). Пацієнти з гострими симптоматичними нападами мали майже вдвічі більший ризик смерті в лікарні порівняно з тими, хто не мав нападів, що свідчить про те, що принаймні для ішемічного інсульту гострі симптоматичні напади дійсно можуть негативно вплинути на результат пацієнта незалежно від тяжкості захворювання [21].
Цереброваскулярні причини
Орієнтовна частота гострих симптоматичних нападів після інсульту становить 3–6 %. Частота нападів значно вища у пацієнтів із геморагічним інсультом (10–18 %), ніж з ішемічним (2–4 %) [22]. Лобарний внутрішньомозковий крововилив, субарахноїдальний крововилив та ішемічний інсульт із вторинною геморагічною трансформацією показують сильніший зв’язок із гострими симптоматичними нападами, ніж ішемічний інсульт [22]. Хоча ішемічний інсульт асоціюється з нижчим ризиком порівняно з внутрішньомозковим крововиливом, на нього припадає більший загальний тягар постінсультних нападів через більшу частоту виникнення. Ішемічний інсульт є провідною етіологією гострих симптоматичних нападів у літніх людей.
Гострі симптоматичні напади зазвичай виникають протягом перших одного-двох днів після церебральної ішемії, приблизно у двох третинах випадків — протягом перших 24 годин. Більшість нападів, пов’язаних з геморагічним інсультом, виникає на початку або протягом перших 24 годин. Більшість нападів є вогнищевими незалежно від типу інсульту або часу виникнення нападу. Це не дивно з огляду на прийняту гіпотезу про те, що вогнищеве пошкодження після інсульту може діяти як вогнище епілептичної активності, а швидке двостороннє поширення від генератора фокального нападу не можна виключити у випадку генералізованих нападів [23].
Вважається, що гострі симптоматичні напади після інсульту є результатом гострої біохімічної дисфункції та вивільнення збуджувальних нейромедіаторів, що призводить до тимчасових змін збудливості нейронів і електрично подразненої тканини [24]. Прозбуджуючі клітинні зміни виникають після гострого ішемічного пошкодження ней-ронів і включають накопичення внутрішньоклітинного кальцію та натрію та підвищення позаклітинних концентрацій глутамату, що може призвести до деполяризації трансмембранного потенціалу та зниження судомного порогу [24]. Рецидивуючі нейронні розряди епілептиформного типу спостерігалися в нейронних мережах нейронів, що вижили, і тимчасові деполяризації відбуваються в ішемічній напівтіні після експериментальної оклюзії середньої мозкової артерії [25].
Менш чітко з’ясований механізм ініціації нападів через кровотечу. Вважається, що продукти крові в паренхімі та похідні метаболізму гему та заліза сприяють фокальному церебральному подразненню. Геморагічна трансформація ішемічного інфаркту мозку пов’язана зі значно вищим ризиком гострих симптоматичних нападів, ніж сам по собі ішемічний інсульт, що додатково підтверджує роль екстравазації крові в розвитку аномальної епілептиформної активності [22].
Ураження кори є добре визначеним фактором ризику ранніх нападів як при ішемічному [19], так і при геморагічному інсульті [26, 27]. Відповідно, стратифікація за підтипом і місцем інсульту має важливе значення для оцінки індивідуального ризику виникнення гострих симптоматичних нападів [25].
У деяких дослідженнях тяжкість неврологічного дефіциту, розмір ураження та молодший вік (< 65 років) також асоціювалися з ранніми нападами. Метаболічні порушення, такі як високий рівень глюкози в крові або низький рівень натрію, можуть спровокувати напади. Вважається, що вживання алкоголю, перенесений інсульт, низький бал Альбертської програми інсульту за ранньою шкалою КТ (ASPECTS) і низький рівень артеріального тиску підвищують ризик гострих симптоматичних нападів після інсульту [26].
Концепція про те, що кардіогенні емболії частіше спричиняють гострі напади, все ще залишається супе-речливою. Визначення кардіоемболічного інсульту та діагностична робота для оцінки джерел серцевої емболії сильно відрізнялися в різних дослідженнях, і часта асоціація серцевої емболії з ураженням кори головного мозку є потенційною перешкодою [25].
Терапія гострого інсульту, зокрема внутрішньовенний тромболізис і механічна тромбектомія, потенційно може вплинути на ризик нападів, збільшуючи або зменшуючи ризик. В експериментальних моделях було показано, що рекомбінантний тканинний активатор плазміногену має різноспрямовані ефекти: з одного боку — нейротоксичний і проконвульсивний ефект, такий як втрата ГАМКергічних інгібуючих інтернейронів, підвищення регуляції матриксних металопротеїназ, пошкодження гематоенцефалічного бар’єра та надмірне вироблення оксиду азоту, з іншого — нейропротекторний ефект і протисудомні властивості, такі як стимуляція нейротрофічного фактора мозку, інгібування апоптозу та стабілізація клітинного енергозабезпечення [30]. Раптові зміни церебральної перфузії та відновлення мозкового кровообігу можуть спровокувати запальний каскад, сприяючи розвитку реперфузійного синдрому та подальших нападів. Однак успішна реканалізація може обмежити ступінь первинного ураження мозку [31].
Проспективне дослідження 516 пацієнтів з ішемічним інсультом не виявило істотної різниці в частоті гострих симптоматичних нападів у тих пацієнтів, які отримували реперфузійну терапію (внутрішньовенне введення рекомбінантного тканинного активатора плазміногену та ендоваскулярна тромбектомія), порівняно з тими пацієнтами, хто її не отримував [32]. Навпаки, у дослідженні типу «випадок — контроль», проведеному в одному центрі лікування інсульту, внутрішньовенний тромболізис був незалежно пов’язаний із появою гострих симптоматичних постінсультних нападів [33]. Наразі недостатньо даних, щоб зробити остаточні висновки, тому фактичний вплив лікування гострого інсульту на ризик нападів залишається незрозумілим [30].
У кількох дослідженнях вивчались аномалії ЕЕГ як предиктори ранніх постінсультних нападів. Латералізовані періодичні розряди (lateralized periodic discharges (LPD)) і фронтальна періодична ритмічна дельта-активність (frontal intermittent rhythmic delta activity (FIRDA)) були виявлені у 25 % і дифузне уповільнення приблизно у 30 % пацієнтів з гострими симптоматичними нападами. Ці аномалії ЕЕГ були значно більш частими у пацієнтів з інсультом із гострими симптоматичними нападами, ніж у пацієнтів без гострих або наступних нападів з пізнім початком [34]. LPD розглядаються як нестабільний нейробіологічний процес з іктально-міжнападовим континуумом, що призводить до маніфестації нападів, коли одночасно існують гострі метаболічні порушення [35]. В експериментальних моделях LPD з’являються в напівтіньових ділянках після оклюзії середньої мозкової артерії в порівнянні з переривчастою ритмічною дельта-активністю, яка виникає в контралатеральній півкулі з лобно-тім’яним домінуванням [36].
Була досліджена кореляція між біомаркерами крові та гострими симптоматичними нападами після інсульту. Серед панелі з 14 біомаркерів, зібраних протягом шести годин після госпіталізації та перед застосуванням будь-якого лікування, було виявлено, що вищі рівні молекулярної адгезії нейронних клітин (NCAM) і нижчі рівні фактора некрозу пухлини рецептора-1 (TNF-R1) є незалежними предикторами раннього початку нападу. NCAM відіграє роль у клітинній адгезії та синаптичній пластичності. TNF-R1 широко бере участь у запальних процесах, і нижчі рівні можуть бути пов’язані зі зв’язуванням із TNF-α, прозапальним цитокіном із проконвульсивним ефектом [37]. Незважаючи на дослідницький характер нашої роботи, комбіноване використання клінічних і електроенцефалографічних змінних із сироватковими біомаркерами може бути цікавою стратегією для виявлення пацієнтів із вищим ризиком ранніх нападів.
Як при ішемічному, так і при геморагічному інсульті гострі симптоматичні напади є прогностичними факторами для розвитку епілепсії [13, 38]. Проте застосування протинападових препаратів, розпочате під час гострих симптоматичних нападів, не змінює ризику розвитку пізньої епілепсії [39].
Церебральний венозний тромбоз пов’язаний з особливо високим ризиком гострих симптоматичних нападів, які є основним симптомом в гострій фазі у 40 % хворих дорослих [40] і третини дітей [1]. Фактори ризику нападів, пов’язаних із церебральним венозним тромбозом, включають супратенторіальні, особливо геморагічні, ураження головного мозку, неврологічні рухові та сенсорні порушення й тромбоз верхнього сагітального синуса та кортикальних вен [41].
Інфекційні причини
Інфекційні захворювання ЦНС є частою етіологією гострих симптоматичних нападів, які виникають приблизно у 20 % пацієнтів з інфекціями ЦНС [42, 43].
Інфекції ЦНС викликають гострі симптоматичні напади як у дорослих, так і у дітей, але спектр інфекцій ЦНС є різним у цих групах (наприклад, бактеріальний менінгіт частіше зустрічається у дітей). У більшості досліджень вивчались гострі симптоматичні напади у зв’язку з інфекціями ЦНС як у дорослих, так і в дітей. Огляд літератури про гострі симптоматичні напади при інфекціях ЦНС також потребує врахування того, що поширеність і спектр інфекцій ЦНС як у дітей, так і у дорослих відрізняються в різних географічних регіонах.
Ризик виникнення гострих симптоматичних нападів є найвищим при вірусному енцефаліті. У групі з 147 дорослих пацієнтів з різними інфекціями ЦНС пацієнти з вірусним енцефалітом мали в 14 разів більше шансів на розвиток нападів, ніж пацієнти з іншими інфекціями ЦНС, такими як бактеріальний менінгіт [43]. У проспективному дослідженні 148 пацієнтів з вірусним енцефалітом гострі симптоматичні напади були зафіксовані у 42,6 % з них [44]. Напади в цьому дослідженні були як фокальними, так і генералізованими, а епілептичний статус розвивався у чверті пацієнтів. Слід зазначити, що епілептичний статус при енцефаліті є більш рефрактерним, ніж при інших етіологіях [45]. Відомості про частоту нападів при енцефаліті, ймовірно, недооцінені, оскільки більш легкі напади та неконвульсивний епілептичний статус можна просто не помітити у пацієнта з симптомами зміненого психічного стану, якщо не застосовувати постійний моніторинг ЕЕГ [46]. Пригнічений рівень свідомості, МРТ-ознаки ураження кори головного мозку та молодший вік є провісниками нападів при вірусному енцефаліті [44]. Напади при енцефаліті пов’язані з поганим прогнозом [44] і підвищеною смертністю [47]. Енцефаліт, спричинений вірусом простого герпесу (HSV) 1-го типу, є найпоширенішою причиною спорадичного енцефаліту та найбільш сильно пов’язаний із гострими симптоматичними нападами у 60 % випадків у гострій стадії [48]. Висока частота нападів, що спостерігається при HSV-енцефаліті, може бути пояснена схильністю HSV інфікувати епілептогенні мезіальні скроневі структури мозку, включаючи гіпокамп [49]. Було показано, що інфікування вірусом герпесу in vitro клітин гіпокампа щурів змінює збудливість клітин гіпокампа та безпосередньо індукує епілептиформну активність [50]. Високі показники гострих симптоматичних нападів також зареєстровані для японського енцефаліту — найпоширенішої форми епідемічного вірусного енцефаліту. У проспективному дослідженні 144 пацієнтів з японським енцефалітом гострі симптоматичні напади виникли у 41 % випадків [47]. Гострі напади при вірусному енцефаліті можуть розвинутися через спричинену вірусом загибель нейрональних клітин, однак запальна реакція на вірусну інфекцію, ймовірно, є більш важливою. Було показано, що цитокіни, як-от TNF-α, IL-1β та IL-6, змінюють синаптичну передачу, що призводить до посилення збудливості нейронів [48]. Вважається, що патофізіологія гострих симптоматичних нападів при вірусному енцефаліті відрізняється від патофізіології пізніх нападів та епілепсії, але вони залишаються недостатньо вивченими. Однак ранні напади при енцефаліті пов’язані з вищим ризиком виникнення пізніх нападів та епілепсії. У дослідженні 714 пацієнтів з енцефалітом 20-річний ризик розвитку неспровокованих нападів становив 22 % для пацієнтів з вірусним енцефалітом і гострими симптоматичними нападами та 10 % для пацієнтів без нападів [42].
Бактеріальний менінгіт є ще однією частою інфекційною причиною гострих симптоматичних нападів у дітей і дорослих. Вважається, що поєднання гнійної запальної реакції та прямого впливу бактеріальних токсинів викликає запалення кори, що призводить до гострих нападів [51]. У проспективному дослідженні 185 дітей з бактеріальним менінгітом у 31 % випадків у них розвинулися гострі симптоматичні напади [52]. Вік до двох років, інфекція Streptococcus pneumoniae, зміна психічного стану та кількість лейкоцитів у спинномозковій рідині нижче 1000 клітин були визначені як незалежні предиктори гострих симптоматичних нападів у дітей з бактеріальним менінгітом [53]. Діти з бактеріальним менінгітом і гострими симптоматичними нападами мають більш високий рівень смертності, ніж діти без нападів [53]. У дорослих з бактеріальним менінгітом про гострі симптоматичні напади повідомлялося в 17–27 % випадків [2, 54, 55]. Факторами ризику виникнення гострих симптоматичних нападів у дорослих із бактеріальним менінгітом є зниження рівня свідомості під час госпіталізації, позитивний посів спинномозкової рідини на Streptococcus pneumoniae, кількість клітин в спинномозковій рідині менше 1000, підвищення рівня d-білка ліквору та вогнищеві аномалії при візуалізації мозку [54]. Як для вірусного енцефаліту, так і для бактеріального менінгіту гірший прогноз мають пацієнти із гострими симптоматичними нападами, ніж пацієнти без них, що свідчить про більшу тяжкість основного захворювання у пацієнтів із гострими симптоматичними нападами.
ГСН часто відбуваються при інших інфекційних захворюваннях ЦНС. До 25 % пацієнтів з абсцесом мозку мають напади [56]. Найчастішим симптомом при нейроцистицеркозі є напади, які виникають у 60–90 % пацієнтів [57]. Напади, які виникають за наявності дегенеративної кісти нейроцистицеркозу з набряком при візуалізації мозку, слід розглядати як гострі симптоматичні напади [1]. Після зникнення гострого запального ураження рецидив нападів не спостерігається у більшості пацієнтів [58]. Напади, які виникають після зникнення запального вогнища та за наявності кальцинованої кісти, слід розглядати як неспровоковані напади, які потребують тривалого протинападового лікування [59].
Автоімунні та запальні причини
Дуже гетерогенна група переважно імуноопосередкованих розладів по-різному пов’язана з гострими симптоматичними нападами і включає ті, що впливають виключно на ЦНС, такі як автоімунний енцефаліт або розсіяний склероз, і ті, які зазвичай викликають системні симптоми з випадковим ураженням ЦНС, як-от системний червоний вовчак (СЧВ).
Існує дуже сильний зв’язок між гострими симптоматичними нападами та неінфекційними імуноопосередкованими формами енцефаліту [60]. Дві групи цих розладів розрізняються за патофізіологічними та клінічними характеристиками — паранеопластичні синдроми [61] та синдроми опосередкованого антитілами автоімунного енцефаліту. При паранеопластичних синдромах імунологічний механізм опосередковується цитотоксичними Т-клітинами, що супроводжується наявністю непатогенних антитіл проти внутрішньоклітинних антигенів. Ризик гострих симптоматичних нападів залежить від залучених ділянок мозку. Такий ризик є особливо високим, якщо залучена лімбічна система [61]. Переважно хронічний і резистентний до лікування запальний процес при цих захворюваннях сприяє стійкій схильності до виникнення нападів, тому у багатьох пацієнтів (до 60 %) розвивається епілепсія [61]. У синдромах опосередкованого антитілами автоімунного енцефаліту імунна відповідь, яка зазвичай має невідому причину, хоча і може бути викликана пухлиною або попереднім вірусним енцефалітом, генерує антитіла, спрямовані проти нейрональних поверхневих антигенів [62]. Захворювання є прямим результатом патогенної взаємодії антитіл з поверхневими антигенами нейронів, і клінічні прояви змінюються залежно від конкретного антигену. Напади часто є провідним симптомом опосередкованого антитілами автоімунного енцефаліту і розвиваються у 33–100 % пацієнтів залежно від антигена-мішені [61]. Автоімунний енцефаліт є найбільш часто виявленою причиною вперше виниклого рефрактерного епілептичного статусу (NORSE) [63]. Антигенами, які найбільше пов’язані з нападами, є рецептор N-метил-D-аспартату (NMDA), рецептори гамма-аміномасляної кислоти (ГАМК) А, В та білок LGI1. Більшість пацієнтів з антитілоопосередкованим автоімунним енцефалітом має сприятливий результат при належному лікуванні за допомогою імунотерапії [62]. Напади зазвичай зникають, коли зникає енцефаліт. Таким чином, напади при опосередкованому антитілами автоімунному енцефаліті, навіть якщо вони виникають протягом тривалого періоду часу, є гострими симптоматичними нападами, оскільки усунення антитіла, що викликає преципітацію, зазвичай зупиняє напади. За винятком рецепторного енцефаліту LGI1 і ГАМК-А, ризик повторних нападів після гострої запальної фази опосередкованого антитілами автоімунного енцефаліту низький (наприклад, < 5 % при енцефаліті рецептора NMDA) [61]. Симптоми нападів у різних підтипах автоімунного енцефаліту не є специфічними, і немає симптомів, які чітко відрізняють імуноопосередковані та неімунні причини [3].
Епілептичні напади виникають приблизно у 15 % пацієнтів із СЧВ [64]. Дослідження 60 пацієнтів із СЧВ з епілептичними нападами показало, що 88 % пацієнтів мали гострі симптоматичні напади і лише 12 % мали повторні напади [65]. Напади виникають здебільшого на початку захворювання або під час його загострення, можуть бути спричинені інсультом на тлі протромботичного стану або васкуліту, інфекції ЦНС, гіпертензії/PRES, ниркової недостатності або, ймовірно, прямої дії на ЦНС антифосфоліпідних антитіл [64].
Інші системні автоімунні розлади, пов’язані з нападами, включають синдром Шегрена, гранулематоз з поліангіїтом, саркоїдоз, целіакію, хворобу Крона та хворобу Бехчета [64]. Існує багато причин нападів при цих розладах, у тому числі прямий імунологічний вплив на мозок через цитокіни, імунні комплекси та автоантитіла, а також судинні захворювання, інфекції, метаболічні розлади та побічні ефекти імуносупресивної терапії [10].
Травма мозку
Гострі симптоматичні напади (протягом одного тижня) клінічно маніфестують після ЧМТ середньої тяжкості у 2–15 % пацієнтів, при цьому більшість досліджень показують частоту близько 3–5 %. Приблизно половина нападів відбувається в перші 24 години [66–68]. Вищі показники нападів спостерігаються при постійному моніторингу ЕЕГ, тобто не всі напади виявляються клінічно. Протинападові препарати, які застосовуються з профілактичною метою після травми голови, можуть зменшити частоту ранніх нападів (перший тиждень), але не впливають на розвиток посттравматичної епілепсії [66, 68]. Найбільш ретельно вивченими протинападовими препаратами є фенітоїн і леветирацетам. Пацієнти з нападами, що маніфестують в момент удару (так звані напади удару, конвульсії від удару), мають низький ризик подальших нападів. Імовірно, такий ризик нижчий, ніж у пацієнтів з іншими ранніми нападами протягом першого тижня після травми [69].
Фактори ризику виникнення гострих нападів після ЧМТ включають більш серйозну травму, необхідність нейрохірургічного втручання, вдавлений перелом черепа, молодший вік (набагато вищий ризик у маленьких дітей, ніж у дорослих), проникне поранення та будь-який тип внутрішньочерепного крововиливу. Як і у випадку з іншими станами, обговореними вище, гострі симптоматичні напади після ЧМТ є основним фактором ризику розвитку епілепсії у метааналізі факторів ризику розвитку посттравматичної епілепсії [70]. В одному дослідженні було встановлено, що гострі епілептиформні аномалії на ЕЕГ були незалежним фактором ризику розвитку посттравматичної епілепсії після тяжкої ЧМТ [71].
Ліки, інтоксикації та алкоголь
Напади, пов’язані з прийомом ліків, можуть виникати внаслідок як прийому, так і відміни певних ліків. У ретроспективному дослідженні 276 пацієнтів із нападами, що виникли вперше, було виявлено, що 6,1 % нападів були пов’язані з ліками та наркотиками [72]. Кокаїнове отруєння, відмова від бензодіазепінів і бупропіону були трьома основними причинами нападів, пов’язаних з ліками та наркотиками. Найпоширенішими ліками, пов’язаними з нападами, є антидепресанти (бупропіон, циталопрам, венлафаксин, триміпрамін, амітриптилін, мапротилін), антипсихотики (клозапін, хлорпромазин, кветіапін), антигістамінні препарати (димедрол) і анальгетики (трамадол і мефенамінова кислота) [73–76]. Однак загальний ризик нападів судом при застосуванні цих ліків, особливо в порівнянні з їх широким використанням, здається низьким і не повинен перешкоджати їх використанню, коли це необхідно. Деякі внутрішньовенні антибіотики мають підвищений ризик виникнення гострих симптоматичних нападів, включаючи цефалоспорини четвертого покоління (особливо цефепім [77]), карбапенеми (особливо іміпенем [78]) і ципрофлоксацин, переважно при застосуванні у високих дозах і пацієнтам з нирковою недостатністю, ураженнями головного мозку або вже наявною епілепсією [79].
Напади, пов’язані зі скасуванням ліків, найчастіше виникають при відміні бензодіазепінів і барбітуратів. Кокаїн і амфетаміни є рекреаційними наркотиками, скасування яких найбільше пов’язане з нападами [80]. Напади потенційно можуть бути спричинені певними галюциногенами, вони навряд чи пов’язані зі зловживанням героїном або марихуаною [1].
Вживання алкоголю є дуже частою причиною гострих симптоматичних нападів. Деякі дослідження показують, що близько третини всіх госпіталізацій, пов’язаних з нападами, спричинені вживанням алкоголю [81]. Найчастіше причиною нападів є алкогольна абстиненція, проте напади можуть виникати і при сильному алкогольному сп’янінні. Напад в алкогольній абстиненції підозрюють у пацієнта з хронічним зловживанням спиртними напоями в анамнезі та нещодавно зменшеним споживанням алкоголю, у якого через 7–48 годин після останнього вживання алкоголю виникає генералізований тоніко-клонічний напад разом із типовими симптомами абстиненції, такими як тремор, пітливість і тахікардія. Вважається, що напади алкогольної абстиненції виникають через гіперзбудливий стан мозку внаслідок змін сигналізації рецепторів NMDA і ГАМК-А, які розвиваються під час хронічного зловживання алкоголем [82]. Необхідна обережність при визначенні нападу як пов’язаного з алкоголем: у ретроспективному дослідженні 140 пацієнтів, у яких спочатку вважали напади пов’язаними з алкоголем, в подальшому у 53,6 % пацієнтів, крім алкоголю, виявили альтернативні причини, такі як травма голови, епілепсія, інсульт та метаболічні аномалії [83]. Європейські фахівці рекомендують візуалізацію мозку та ЕЕГ навіть в очевидних випадках першого нападу, пов’язаного з алкоголем [81].
Еклампсія
Еклампсія може маніфестувати розвитком гострих симптоматичних епілептичних нападів у жінки під час вагітності або після пологів, яка має ознаки та симптоми прееклампсії, як-от гіпертензія, протеїнурія та набряки [84]. Еклампсія є рідкісним ускладненням вагітності з 1,5–10 випадками на 10 000 пологів і несе значний ризик тяжкої материнської захворюваності та смертності [85]. Приблизно половина нападів виникає перед пологами, але напади можуть розвинутися під час пологів і в перші дні після них [86]. Напади є фокальними або двосторонніми тоніко-клонічними, їм часто передують головний біль і розлади зору. Такі напади зазвичай розвиваються у пацієнток, які вже мали ознаки прееклампсії. Однак напади можуть розвинутися у пацієнток до маніфестації гіпертензії та протеїнурії [87]. Церебральна МРТ демонструє гіперінтенсивні ураження білої речовини в режимі T2 з тім’яно-потиличним домінуванням [88]. Лікування включає призначення сульфату магнію, який значно ефективніше знижує ризик повторних нападів, ніж діазепам і фенітоїн. Сульфат магнію також ефективний для первинної профілактики нападів у пацієнток з прееклампсією [89].
Церебральна аноксія
Аноксія внаслідок серцево-легеневої зупинки часто спричиняє церебральне пошкодження, яке залучає таламус, гіпокамп і кіркові пірамідні клітини і може призвести до нападів [90]. Напади, що маніфестують клінічно, виникають у третини коматозних пацієнтів з аноксичною енцефалопатією [91]. У більшості розвивається як кортикальний, так і/або підкірковий міоклонус, тоді як генералізовані тоніко-клонічні напади виникають лише у 7 % усіх пацієнтів з аноксичною енцефалопатією. Міоклонії прогресують до міоклонічного епілептичного статусу у меншості пацієнтів. Повідомлялося про неконвульсивний епілептичний статус у третини коматозних пацієнтів з аноксичною енцефалопатією [92]. Міоклонії/міоклонічний епілептичний статус та неконвульсивний епілептичний статус довго розглядалися як предиктори поганого неврологічного результату, однак у новітній літературі припускається, що хороший неврологічний результат все ще можливий [90]. Пацієнтів з тоніко-клонічними нападами й аноксичною енцефалопатією слід лікувати протинападовими препаратами, як і в разі будь-якого іншого гострого розладу. Бензодіазепіни, вальпроєва кислота та леветирацетам можуть бути найбільш ефективними при міоклонічних нападах [93]. Немає єдиної думки щодо того, наскільки агресивно слід лікувати пацієнтів з неконвульсивним епілептичним статусом з аноксичною енцефалопатією через відсутність доказової бази [94]. Однак з’являється все більше повідомлень про зв’язок успішного лікування пацієнтів з постаноксичним рефрактерним неконвульсивним епілептичним статусом та хорошим неврологічним результатом, що обґрунтовує більш агресивне та тривале лікування протинападовими препаратами та седативними засобами [95].
Менеджмент
Для успішного лікування гострих симптоматичних нападів необхідне чітке розуміння того, що напад є саме гострим симптоматичним і вжито негайних діагностичних заходів для виявлення основної патології, яка призвела до його розвитку. Інформація з анамнезу пацієнта та результати фізикального та неврологічного обстеження призведуть до можливого розуміння того, що напад може мати чітку етіологічну причину, тобто є саме гострим симптоматичним. Потім ще слід провести лабораторні дослідження та візуалізацію мозку, а також ЕЕГ-дослідження. Якщо є можливість, за показаннями доцільне проведення люмбальної пункції. Після встановлення діагнозу слід якнайшвидше призначити лікування основного захворювання (наприклад, внутрішньовенний тромболізис, механічна тромбектомія при ішемічному інсульті, ацикловір при герпетичному енцефаліті, антикоагулянт при тромбозі церебральних вен тощо).
Пацієнтів із гострими симптоматичними нападами слід лікувати протинападовими препаратами під час гострої фази основного захворювання або доки присутній провокуючий фактор або причина таких нападів. У пацієнтів з гострими симптоматичними нападами можуть розвинутися подальші гострі симптоматичні напади незабаром після першої події [96]. Метою лікування протинападовими препаратами є запобігання подальшим гострим симптоматичним нападам. Однак даних про те, як довго лікувати пацієнтів із гострими симптоматичними судомами за допомогою протинападових препаратів, значною мірою бракує. Ймовірно, тривалість лікування протинападовими препаратами має бути індивідуальною для кожного пацієнта та залежатиме від таких факторів, як основна етіологія (наприклад, повністю вилікувана гіпонатріємія проти триваючого автоімунного енцефаліту), ризик подальших нападів, тяжкість гострого симптоматичного нападу (наприклад, епілептичний статус, травми, пов’язані з нападами), загальна клінічна ситуація (наприклад, чи пацієнт прийшов до тями і почувається добре, чи він все ще перебуває у критичному стані/у реанімації, чи є у нього серйозні супутні захворювання) і користь для пацієнта. Для хворих із гострим структурним ураженням головного мозку можна екстраполювати літературу про ЧМТ і дійти висновку, що лікування протягом одного-двох тижнів протинападовими препаратами знизить частоту ранніх нападів.
Початкове лікування пацієнтів з тяжкою ЧМТ включає в комплекс заходів протинападові препарати, зокрема леветирацетам 1500 мг в/в під час первинного обстеження та протягом 30 хвилин після прибуття у шпиталь, потім 1000 мг двічі на добу; препарат другої лінії лакозамід 400 мг, потім 200 мг двічі на добу; препарат третьої лінії фенітоїн 20 мг/кг одноразово, потім 300 мг на добу.
Профілактичне лікування продовжується протягом 7 днів після помірної або тяжкої ЧМТ. За наявності судомної активності лікування триває після 7-го дня. Застосовують препарати невідкладної допомоги — лоразепам 1–2 г в/в або мідазолам 5–10 мг в/в.
Треба відзначити, що таке лікування застосовується для запобігання розвитку негайних і ранніх нападів та покращення результатів лікування гострої черепно-мозкової травми в бойових умовах (можливо, при тяжкій ЧМТ мирного часу). Застосування протинападових препаратів у гострому періоді ЧМТ не є методом профілактики розвитку посттравматичної епілепсії, тому тривале лікування навряд чи буде ефективним і може призвести тільки до розвитку побічних ефектів протинападових препаратів [97].
Це логічний і, ймовірно, найбільш заснований на доказах підхід, який зараз здійсненний. Однак у багатьох клінічних ситуаціях може знадобитися лікування протинападовими препаратами довше двох тижнів, тому під час прийняття рішення потрібне клінічне судження, яке враховує індивідуальні фактори пацієнта. Тривале протинападове лікування, як правило, не показане, оскільки ризик рецидиву гострих симптоматичних нападів значно нижчий, ніж неспровокованих нападів, як уже обговорювалося вище [7]. Однак, на жаль, у клінічній практиці ми стикаємося з тим, що багатьох пацієнтів залишають на лікуванні протинападовими препаратами протягом відносно тривалих періодів, а часто і на невизначений термін.
Доцільним є проведення ЕЕГ через два-три місяці після виписки з лікарні для тих, хто продовжує прий-мати протинападові препарати через гострі симптоматичні напади або мав значні епілептиформні зміни на ЕЕГ під час гострого процесу. Якщо ЕЕГ не показує епілептиформних розрядів, дозування протинападових препаратів можна зменшувати. Повільне зниження дози протинападового препарату може зменшити ризик виникнення нападів унаслідок саме припинення прийому протинападових препаратів. ЕЕГ доцільно повторювати після відміни протинападових препаратів, особливо якщо пацієнта лікували бензодіазепінами або леветирацетамом, які можуть приховати спайкові зміни. Цей підхід є прагматичним, але не базується на доказах. Якщо можливо, пацієнт повинен брати участь у процесі прий-няття рішення щодо тривалості лікування протинападовими препаратами. Це включає пояснення пацієнту, що ризик повторного нападу після гострого симптоматичного нападу загалом низький, але в деяких ситуаціях може існувати більший ризик подальших нападів.
Висновки
Таким чином, напади, які виникають у тісному часовому зв’язку з ураженням головного мозку і не повторюються після усунення патологічного стану або усунення фактора, що спричинив напад, слід розглядати як гострі симптоматичні напади.
Гострі симптоматичні напади відрізняються від неспровокованих нападів та епілепсії, оскільки ризик рецидиву нападу значно нижчий і немає тривалої схильності до подальших нападів.
Найважливішими причинами гострих симптоматичних нападів у дорослих є як захворювання, що викликають структурні ураження мозку, такі як ішемічний інсульт, крововилив у мозок, травма головного мозку або енцефаліт, так і фактори, які не впливають на структурну цілісність мозку, як-от метаболічні порушення та інтоксикації.
Напади вважаються гострими симптомами, якщо вони виникають протягом 24 годин за наявності серйозного метаболічного розладу, протягом семи днів після гострого структурного ураження мозку, як-от цереброваскулярна подія чи черепно-мозкова травма, або довше, якщо доведено, що процес, який порушує цілісність ЦНС (наприклад, запальні ураження на зображеннях мозку або антитіла до рецепторів NMDA у спинномозковій рідині), триває.
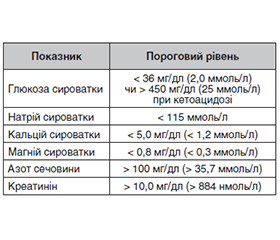
Для гострих симптоматичних нападів при метаболічних розладах були запропоновані граничні значення, згідно з якими причинно-наслідковий зв’язок між судомами та метаболічним розладом вірогідний (наприклад, сироватковий натрій < 115 мг/дл).
У випадках ГСН, викликаних деструктивними ураженнями головного мозку, пацієнти мають вищий ризик розвитку епілепсії, але у більшості цих пацієнтів ніколи не буде неспровокованих нападів.
ГСН виникають рідше, ніж неспровоковані напади. Частота їх виникнення залежить від частоти появи основної патології.
Деякі захворювання, такі як вірусний або автоімунний енцефаліт, церебральний венозний тромбоз або синдром задньої оборотної енцефалопатії, пов’язані з особливо високим ризиком гострих симптоматичних нападів.
Пацієнти з гострими симптоматичними нападами мають високий ризик смертності в перші тижні після події. Ризик смертності переважно опосередковується тяжкістю основного захворювання, але ГСН також може мати незалежний негативний вплив на результат лікування пацієнта.
Пацієнтів із ГСН слід лікувати протинападовими препаратами під час гострої фази основного захворювання, оскільки це може запобігти наступним гострим симптоматичним нападам. Лікування повинно зосереджуватись на терапії відповідного основного захворювання та корекції або усуненні умов або факторів, що провокують напад.
Довготривале лікування протинападовими препаратами, як правило, не є необхідним, і ПНП слід поступово скасовувати протягом тижнів або місяців після гострого симптоматичного нападу.
UA-LEVI-PUB-012025-065
Отримано/Received 12.12.2024
Рецензовано/Revised 24.01.2025
Прийнято до друку/Accepted 29.01.2025

/45.jpg)