
Международный неврологический журнал Том 21, №1, 2025
Вернуться к номеру
Спінальна м’язова атрофія з початком у дорослому віці в пацієнта з мутацією SOD1: клінічний випадок
Авторы: M.B. Mykhailova, M.M. Prokopiv, T.I. Illyash
Bogomolets National Medical University, Kyiv, Ukraine
Рубрики: Неврология
Разделы: Справочник специалиста
Версия для печати
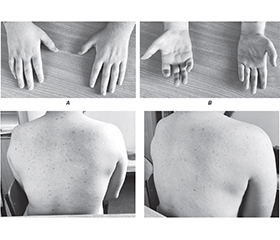
Бічний аміотрофічний склероз (БАС) та спінальна м’язова атрофія (СМА) є одними з найпоширеніших генетично обумовлених захворювань, які характеризуються ураженням мотонейронів. Для БАС типовим є прогресуюче ураження як верхніх, так і нижніх мотонейронів, тоді як СМА переважно вражає нижні мотонейрони. Однак іноді генетичні мутації, зокрема у гені SOD1, що зазвичай пов’язані з БАС, можуть викликати атипові прояви, ускладнюючи диференціальну діагностику. У цьому клінічному випадку 28-річний пацієнт звернувся зі скаргами на прогресуючу дистальну м’язову слабкість, атрофію та степажну ходу, без ознак ураження верхніх мотонейронів. Електроміографія підтвердила хронічну дисфункцію нижніх мотонейронів, що викликало підозру на СМА. Генетичне тестування не виявило делецій у гені SMN1, проте було ідентифіковано патогенну мутацію в гені SOD1. Однак клінічний перебіг захворювання, а саме повільне прогресування симптомів та відсутність ураження верхніх мотонейронів, був більш характерний для СМА IV типу з початком у дорослому віці, а не БАС. Цей випадок демонструє складність діагностики уражень мотонейронів при атипових клінічних і генетичних ознаках. Таким чином, підкреслюється необхідність глибокого генетичного аналізу й мультидисциплінарного підходу для точного встановлення діагнозу та кращого розуміння ролі мутацій SOD1 у формуванні фенотипів, схожих на СМА.
Amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA) are distinct motor neuron disorders with overlapping molecular mechanisms. ALS involves progressive upper and lower motor neuron degeneration, while SMA primarily affects lower motor neurons. Mutations in SOD1 (superoxide dismutase 1), a well-established cause of familial ALS, have been identified in patients with atypical motor neuron disease phenotypes, suggestive of their broader role in motor neuron dysfunction. A 28-year-old male presented with progressive distal muscle weakness, atrophy, and a steppage gait, without upper motor neuron involvement. Electromyography confirmed chronic motor neuron dysfunction, raising suspicion of SMA. Genetic testing excluded SMN1 deletions but identified a pathogenic SOD1 mutation. Despite this, slow disease progression and phenotype were more consistent with adult-onset SMA type IV than ALS. This case highlights the diagnostic challenges posed by overlapping features of ALS and SMA. The findings emphasize the need for further research into the clinical features associated with SOD1 mutations and their potential contributions to SMA-like presentations, refining our understanding of motor neuron disorders.
хвороба мотонейронів; бічний аміотрофічний склероз; спінальна м’язова атрофія; SOD1
motor neuron disease; amyotrophic lateral sclerosis; spinal muscular atrophy; SOD1